세 번째 수업: 한 샘플의 발현량 행렬 읽기: 실습 리뷰와 아웃라이어 유전자 탐색
This content is not available in your language yet.
두 번째 수업에서 우리는 조직에서 뽑은 RNA가 FASTQ → 품질관리 → 트리밍 → 정렬 → 정량(TPM) 을 거쳐 발현량 행렬이 되는 파이프라인을 개념으로 훑었습니다. 이번 수업은 그 파이프라인을 실제로 돌린 결과물을 로그·파일 단위로 리뷰하고, 거기서 나온 딱 한 명의 발현량 벡터 하나로 무엇을 할 수 있는지, 비정상적으로 튀는 유전자를 어떻게 골라내는지, 를 다룹니다.
오늘의 목표: ① 실습에서 실제로 쓴 STAR·RSEM·featureCounts 명령과 결과 파일을 열어 “잘 만들어진 데이터인지” 판별하는 법을 익히고, ② 샘플이 하나뿐일 때 공개 레퍼런스(GTEx·PCAWG)와 나란히 놓고 아웃라이어 유전자를 찾는 지표(백분위·z-score·fold change·max 비교)와 기능 분석까지 한 번에 꿴다. 지난 시간이 데이터 생성 편이었다면, 이번 시간은 RNA analysis (2): 해석 편입니다.
1. 실습 파이프라인 한눈에
섹션 제목: “1. 실습 파이프라인 한눈에”지난 시간에 개념으로 본 순서를, 이번엔 실제 명령을 붙여 끝까지 돌렸습니다.
FastQC → Trim Galore! → FastQC → STAR → (RSEM · featureCounts) → 후반 분석
- 앞의 다섯 단계가 발현량 행렬을 만드는 과정,
- 마지막 후반 분석이 이번 수업의 본론: 만들어진 행렬을 공개 데이터와 비교해 특이 신호를 잡는 과정입니다.
FastQC: 트리밍 전/후, 그리고 두 회사 데이터
섹션 제목: “FastQC: 트리밍 전/후, 그리고 두 회사 데이터”이번 샘플은 같은 T0 종양을 두 시퀀싱 회사(Tempus, BostonGene)에 각각 맡긴 데이터입니다: 같은 검체를 재현성·검증 목적으로 두 곳에 보낸 것으로 보입니다.
FastQC를 트리밍 전(왼쪽)·후(오른쪽) 로 각각 돌려 비교했더니:
- 트리밍 후 total read 수가 소폭 감소: 너무 짧은 read가 걸러졌기 때문.
- 말단(3′ 끝) 품질이 개선: 품질 낮은 꼬리 부분이 잘려 나가서.
- 원래 품질이 좋았고 어댑터 오염도 크지 않아, 이 데이터는 큰 보정 없이 통과. (일부 데이터에서 흔히 보이는 read 앞부분 어댑터 절단 이슈는 여기선 없었습니다.)
요지: FastQC의 결과는 “몇 점 이상 통과”가 아니라, 트리밍이 의도대로 저품질·어댑터를 걷어냈는지를 눈으로 확인하는 용도입니다. → 각 항목을 어떻게 읽는지는 QC(FastQC) 레퍼런스.
2. STAR: 실제로는 이런 명령을 썼다
섹션 제목: “2. STAR: 실제로는 이런 명령을 썼다”지난 시간 슬라이드의 STAR 명령은 최소 입력만 담은 단순한 형태였습니다:
STAR --genomeDir ./ref \ --readFilesIn sample_R1_val_1.fq.gz sample_R2_val_2.fq.gz \ --runThreadN 8 --outSAMtype BAM SortedByCoordinate그런데 실제 Sid 케이스의 로그를 참고하니 훨씬 더 정교한 옵션 세트를 쓰고 있었습니다. 핵심만 추리면:
STAR --runMode alignReads --runThreadN 16 \ --genomeDir .../STAR_genome_GRCh38_noALT_noHLA_noDecoy_v47_oh75 \ --readFilesIn boston/..._1_val_1.fq.gz boston/..._2_val_2.fq.gz \ --readFilesCommand zcat \ --twopassMode Basic \ # 새 splice junction 탐지 향상 --quantMode TranscriptomeSAM GeneCounts \ # RSEM용 transcriptome BAM 생성 --chimSegmentMin 15 --chimJunctionOverhangMin 15 \ # fusion transcript 탐지 --chimOutType Junctions WithinBAM SoftClip \ --alignSoftClipAtReferenceEnds Yes \ # breakpoint·fusion read 정렬 향상 --outFilterMultimapNmax 20 --outFilterMismatchNoverLmax 0.1 \ --alignIntronMin 20 --alignIntronMax 1000000 \ --outSAMtype BAM Unsorted --outSAMunmapped Within대부분은 정렬 정확도를 높이는 조절값(mismatch 허용 비율, intron 길이 범위, score 최소 기준 등)이라, 옵션마다 논문을 찾을 필요는 없고 STAR 매뉴얼에 각 옵션의 기본값과 역할이 정리돼 있습니다. 다만 의미상 꼭 짚어야 할 네 가지가 있습니다:
| 옵션 | 목적 |
|---|---|
--twopassMode Basic | 1차 정렬에서 발견한 splice junction을 참고해 2차로 다시 정렬 → 새 junction 탐지 향상 |
--chim* (chimeric 계열) | fusion transcript 탐지: 종양 조직에는 insertion·deletion 등 구조 변이가 많아, 서로 다른 유전자가 이어 붙은 융합 전사체가 생김. 이를 잡기 위한 종양 특이 옵션 |
--quantMode TranscriptomeSAM | 뒤에서 RSEM이 TPM을 계산할 때 필요한 transcriptome 정렬 BAM을 추가로 생성 |
--alignSoftClipAtReferenceEnds Yes | breakpoint·fusion read의 끝부분을 soft-clip 허용 → 정렬 향상 |
STAR가 내놓는 것: 두 개의 BAM과 로그
섹션 제목: “STAR가 내놓는 것: 두 개의 BAM과 로그”STAR는 중간 파일을 여럿 만들지만, 실제로 뒤에서 쓰이는 핵심 산출물은 세 가지입니다:
| 파일 | 내용 | 다음 단계 |
|---|---|---|
~.Aligned.out.bam | 게놈에 정렬된 기본 결과: read ID가 몇 번 염색체 어느 위치에 붙었는지 | → featureCounts |
~.Aligned.toTranscriptome.out.bam | 전사체(transcriptome)에 정렬된 결과 | → RSEM |
~.Log.final.out | 정렬 품질 요약 통계 | → 품질 확인 |
💡 RSEM 단계에서 막힌다면 여기부터 의심하세요. RSEM은 게놈 BAM이 아니라
Aligned.toTranscriptome.out.bam을 입력으로 받습니다. STAR에--quantMode TranscriptomeSAM을 주지 않으면 이 파일이 아예 안 만들어져서, RSEM이 넘어가지 않습니다. (실습 중 RSEM에서 하루 종일 헤맨 사례가 바로 이 지점일 가능성이 큽니다.)
STAR 로그 읽는 법: “잘 정렬됐나?”
섹션 제목: “STAR 로그 읽는 법: “잘 정렬됐나?””정렬이 잘 됐는지는 Log.final.out 요약으로 판단합니다.
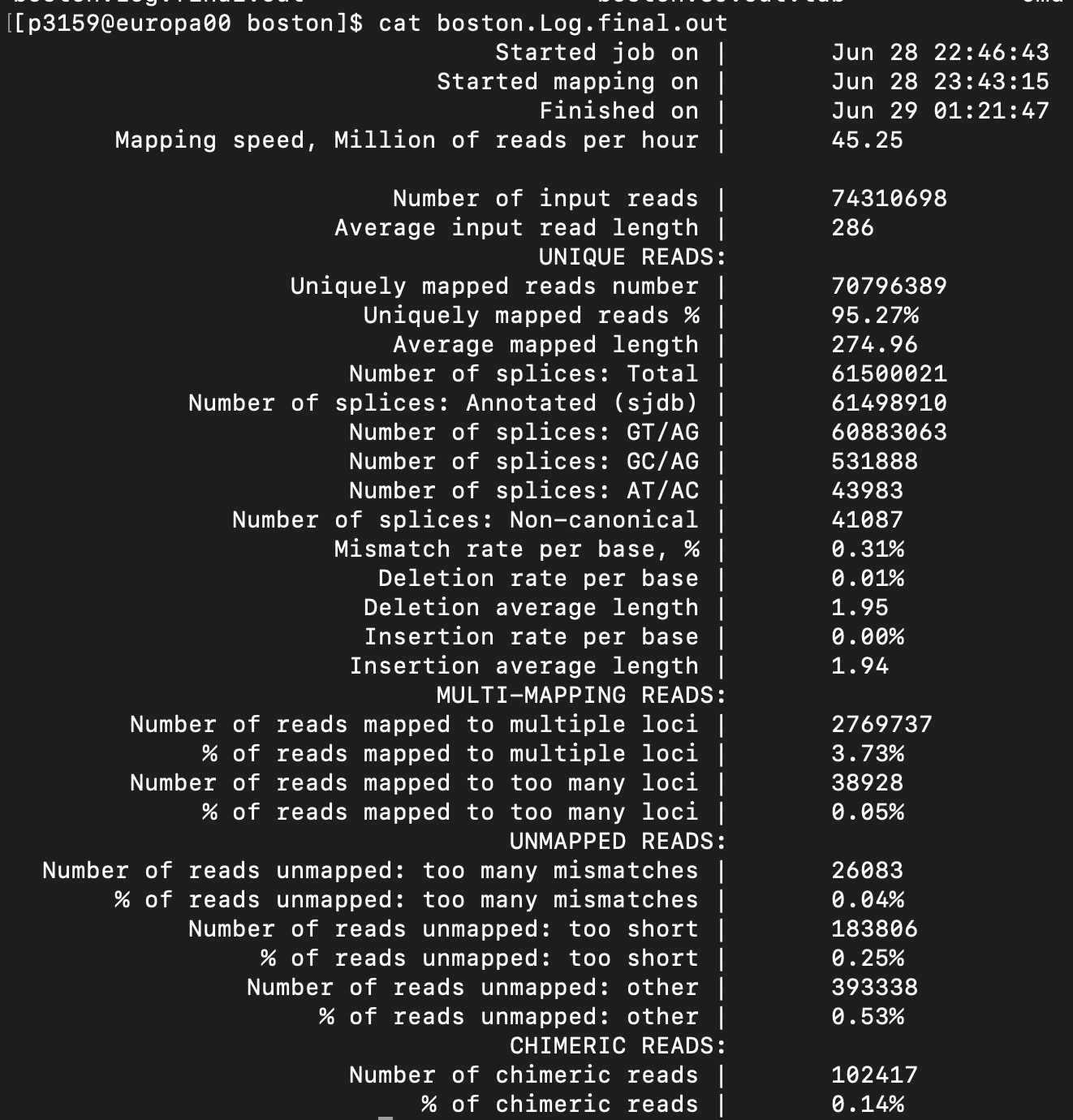
이 실습 샘플(BostonGene)의 STAR 요약. 입력 read 약 7,431만 개 중 Uniquely mapped 95.27%, multiple loci 3.73%, too-short unmapped 0.25%. splice·mismatch·chimeric 통계도 함께 보고된다.
주로 보는 세 수치:
| 지표 | 좋은 기준 | 이 데이터 |
|---|---|---|
| Uniquely mapped reads % | 보통 ≥ 80% 면 양호 | 95.27%: read 대부분이 게놈의 한 자리에 깔끔히 붙음 |
| Multi-mapping % | RNA-seq에서 5~15% 는 정상 | 3.73%: 오히려 낮음 |
| Unmapped % | < 5% 권장 | 합쳐도 1% 미만 |
- Uniquely mapped가 높다는 건, read가 유래한 게놈 위치를 모호함 없이 특정했다는 뜻입니다. 이 데이터는 95% 이상이라 매우 깨끗합니다.
- 만약 이 수치들이 나쁘게 나오면 → 트리밍을 조절해 대응합니다. 과하게 잘라 정렬이 안 되면 덜 자른 버전으로, 저품질이 남아 안 붙으면 더 자르는 식으로.
⏱️ 트리밍을 바꾸면 정렬을 처음부터 다시 해야 합니다. read 세트가 바뀌면 재정렬이 불가피하고, 이 데이터에서 정렬 한 번에 2~5시간이 걸렸습니다. 스레드(
--runThreadN)를 늘리면 빨라지지만 장비 코어 수에 한계가 있습니다.
3. RSEM: 유전자별 TPM과 아이소폼
섹션 제목: “3. RSEM: 유전자별 TPM과 아이소폼”RSEM은 transcriptome BAM을 받아 TPM을 계산하는 도구입니다. 정확도 향상 옵션(--estimate-rspd 등)을 붙여 실행하면 두 개의 결과 파일이 나옵니다.
rsem-calculate-expression --alignments --paired-end \ --estimate-rspd --append-names -p 16 \ boston.Aligned.toTranscriptome.out.bam \ .../rsem_reference_GRCh38_gencode47/rsem_reference boston~.genes.results: 유전자 수준 결과. 유전자마다 expected_count(read 카운트에 해당) · TPM · FPKM을 계산해 줍니다.
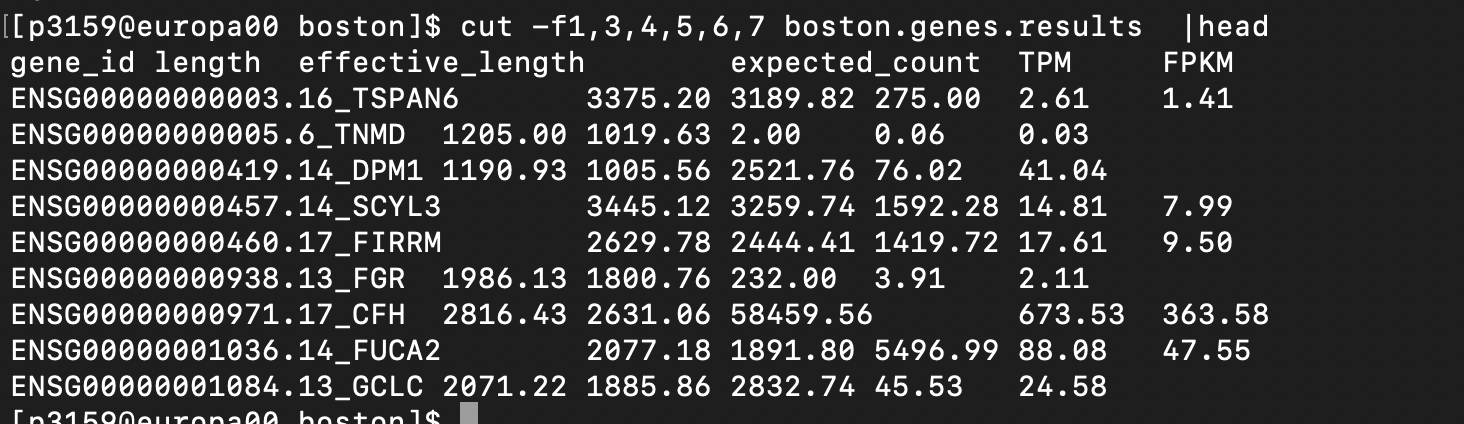
RSEM genes.results. 가운데 transcript_id(s) 컬럼이 그 유전자에 속한 모든 트랜스크립트를 콤마로 이어 붙여 놓아 길고 지저분해 보이므로, 여기서는 cut으로 그 컬럼을 잘라 gene_id · expected_count · TPM · FPKM만 보여준다.
~.isoforms.results: 트랜스크립트(아이소폼) 수준 결과. 같은 유전자라도 개별 아이소폼별로 카운트를 나눠 줍니다.
4. featureCounts: 나중을 위한 raw count
섹션 제목: “4. featureCounts: 나중을 위한 raw count”TPM(RSEM) 은 지금처럼 한 샘플을 공개 레퍼런스와 비교할 때 쓰고, featureCounts 는 raw count(정규화 전 원시 카운트) 행렬을 만드는 용도입니다.
featureCounts -T 16 -p -t exon -g gene_id --extraAttributes gene_name \ -a gencode.v47.annotation.gtf \ -o boston.featureCounts.txt boston.Aligned.out.bam
# 헤더·주석 정리해서 gene_id · gene_name · count 3컬럼으로grep -v "^#" boston.featureCounts.txt \ | awk 'BEGIN{OFS="\t"} NR==1{print "gene_id","gene_name","count"} \ NR>1{print $1,$7,$NF}' > boston.gene_counts.tsv- 입력이 RSEM과 다릅니다. featureCounts는 게놈 정렬 BAM(
Aligned.out.bam)과 GTF(유전자 위치 파일)를 받아, 각 gene_id에 속한 엑손 위치에 붙은 read를 셉니다. 별도 인덱스를 만들 필요 없이 GTF를 바로 씁니다. - 지금 당장은 raw count를 쓰지 않지만, 나중에 차등발현(DEG) 분석을 할 때 필요합니다: DESeq2 같은 도구가 “raw count를 달라” 고 못 박아 두었기 때문입니다(정규화를 자기가 직접 하려고).
💡 정리:
Aligned.out.bam→ featureCounts → raw count,Aligned.toTranscriptome.out.bam→ RSEM → TPM. 같은 STAR 결과지만 어느 BAM을 먹이느냐로 갈립니다. → 발현량 행렬 형식
여기까지가 지난 시간에 예고한 “read에서 발현량 행렬까지” 를 실제로 완주한 부분입니다. 이제 그 행렬로 무엇을 할지가 남았습니다.
5. 후반 분석: 샘플이 딱 하나일 때
섹션 제목: “5. 후반 분석: 샘플이 딱 하나일 때”지금까지 만든 것은 환자 한 명의 종양 조직 RNA-seq 프로파일 하나 입니다. 유전자 수만 개(주석 기준 약 6만 개) 각각의 발현량(TPM 또는 raw count)이 담긴 벡터 하나.
그래서… 이 중 뭐가 중요한가? 발현량 6만 줄을 눈으로 다 볼 수는 없습니다. 필요한 질문은 하나로 좁혀집니다:
“이 종양에서, 정상 조직이나 다른 암과 비교했을 때, 비정상적으로 발현이 튀는 유전자는 무엇인가?”
이 질문에 답하는 작업이 아웃라이어 유전자(outlier gene) 탐색입니다.
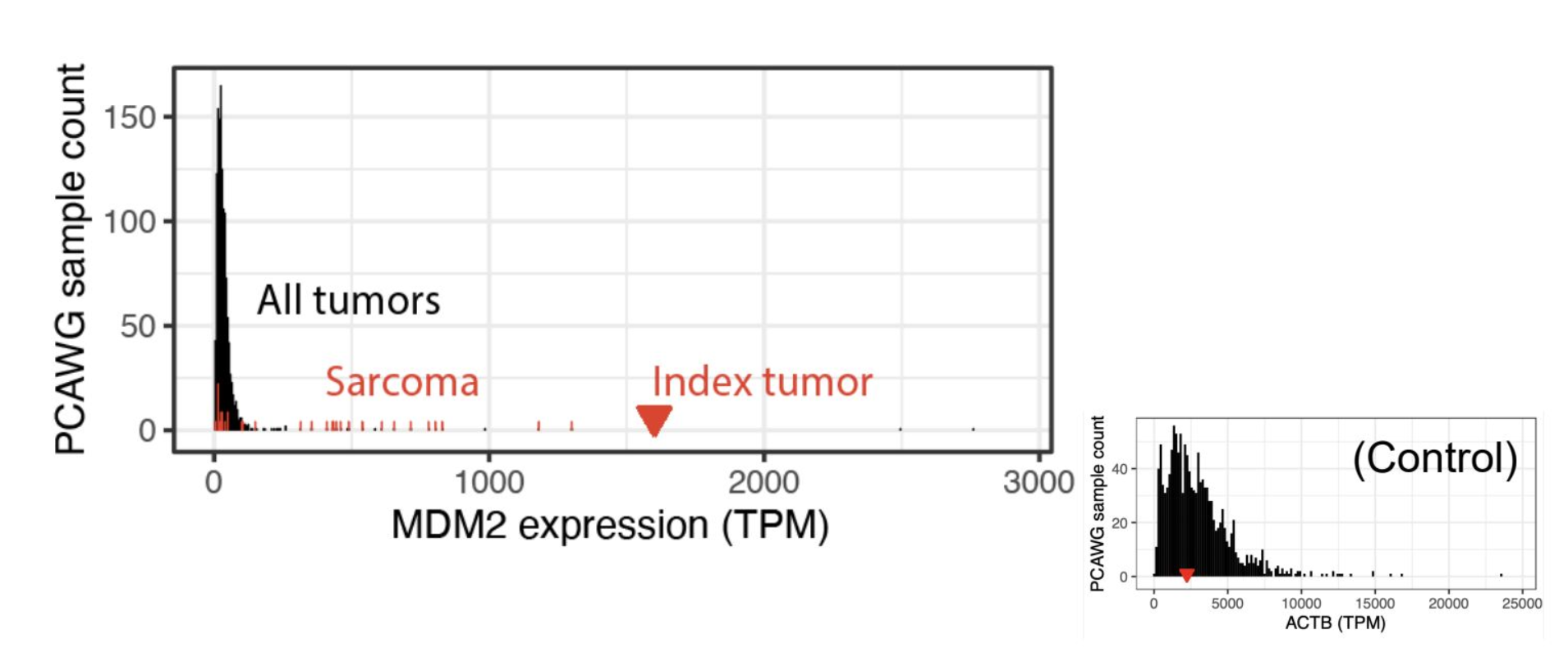
아이디어. 가로축은 한 유전자의 발현량(TPM), 세로축은 그 발현량을 가진 샘플 수. 대부분의 종양(All tumors)은 MDM2가 낮게 몰려 있는데, 이 환자(Index tumor) 는 저 멀리 오른쪽: 육종(Sarcoma) 샘플들과 비교해도 튄다. 오른쪽 작은 그림의 ACTB(housekeeping 유전자)는 대조로, 누구에게나 높아 분포 한가운데 위치한다: 이런 건 아웃라이어가 아니다.
튄다고 곧 원인은 아니다: 해석의 함정
섹션 제목: “튄다고 곧 원인은 아니다: 해석의 함정”발현량이 높게 튀는 걸 발견해도, 그 자체로 “이 유전자가 암을 일으켰다”고 말할 수는 없습니다. 높은 발현은 다음 중 무엇이든 될 수 있습니다:
| 성격 | 의미 |
|---|---|
| 원인(cause) | 종양 발생·진행에 직접 기여 |
| 결과(consequence) | 종양 관련 pathway가 켜지면서 부차적으로 함께 오른 발현 |
| 상태(state) | 종양 미세환경 변화(저산소·영양 부족·전이 능력 획득 등)에 적응하며 바뀐 발현 |
지금 데이터에서 확실히 아는 건 “발현이 튄다” 뿐입니다. 인과를 밝히려면 아래가 함께 필요합니다:
WGS 변이 분석 + pathway 분석 + 문헌조사: 예컨대 과발현이 이 유전자 자체의 변이 때문인지, 아니면 변이는 없는데 다른 조절 유전자의 영향으로 튀는 것인지를 가르고, 후자라면 그 조절 고리를 끊는 접근을 생각해 볼 수 있습니다. 여기에 다른 오믹스(예: 전사인자·효소가 붙을 수 있는 열린 자리를 보는 ATAC-seq)를 얹어 층층이 확인합니다.
그럼에도 아웃라이어 탐색이 유용한 이유는, 정상·타 환자 대비 변화가 분명한 유전자를 추려 표적 치료 후보·후보 약물 탐색의 출발점으로 삼을 수 있기 때문입니다: Sid 케이스가 MDM2 과발현을 표적으로 잡은 것처럼.
6. 레퍼런스 데이터와 검증
섹션 제목: “6. 레퍼런스 데이터와 검증”한 샘플을 “비교”하려면 기준이 될 공개 레퍼런스가 필요합니다. Sid가 쓴 것과 같은, 바로 받아 쓸 수 있는 TPM 수준의 두 데이터를 썼습니다:
| 레퍼런스 | 규모 | 성격 |
|---|---|---|
| GTEx | 74,628 genes · 19,616 샘플 (31 조직) | 정상 조직 발현 |
| PCAWG (EBI 경유) | 56,717 genes · 1,350 샘플 (27 암종) | 다른 암 환자 발현 |
세 데이터셋(내 샘플 + GTEx + PCAWG)에 모두 존재하는 공통 유전자 45,698개만 추려서 비교했습니다.
🧩 실습에서 암 레퍼런스는 EBI(유럽 생물정보학 연구소) 의 Expression Atlas에서 받은 PCAWG 데이터를 써서, 그림·표에
ebi라는 라벨로 표기했습니다. 지난 수업에서 소개한 PCAWG와 같은 계열입니다.
검증 ①: Boston vs Tempus 상관
섹션 제목: “검증 ①: Boston vs Tempus 상관”본론에 들어가기 전에, 만든 count가 믿을 만한지 먼저 봅니다. 같은 검체를 두 회사에 맡긴 데이터이므로, 둘의 발현량은 비슷한 분포여야 합니다.
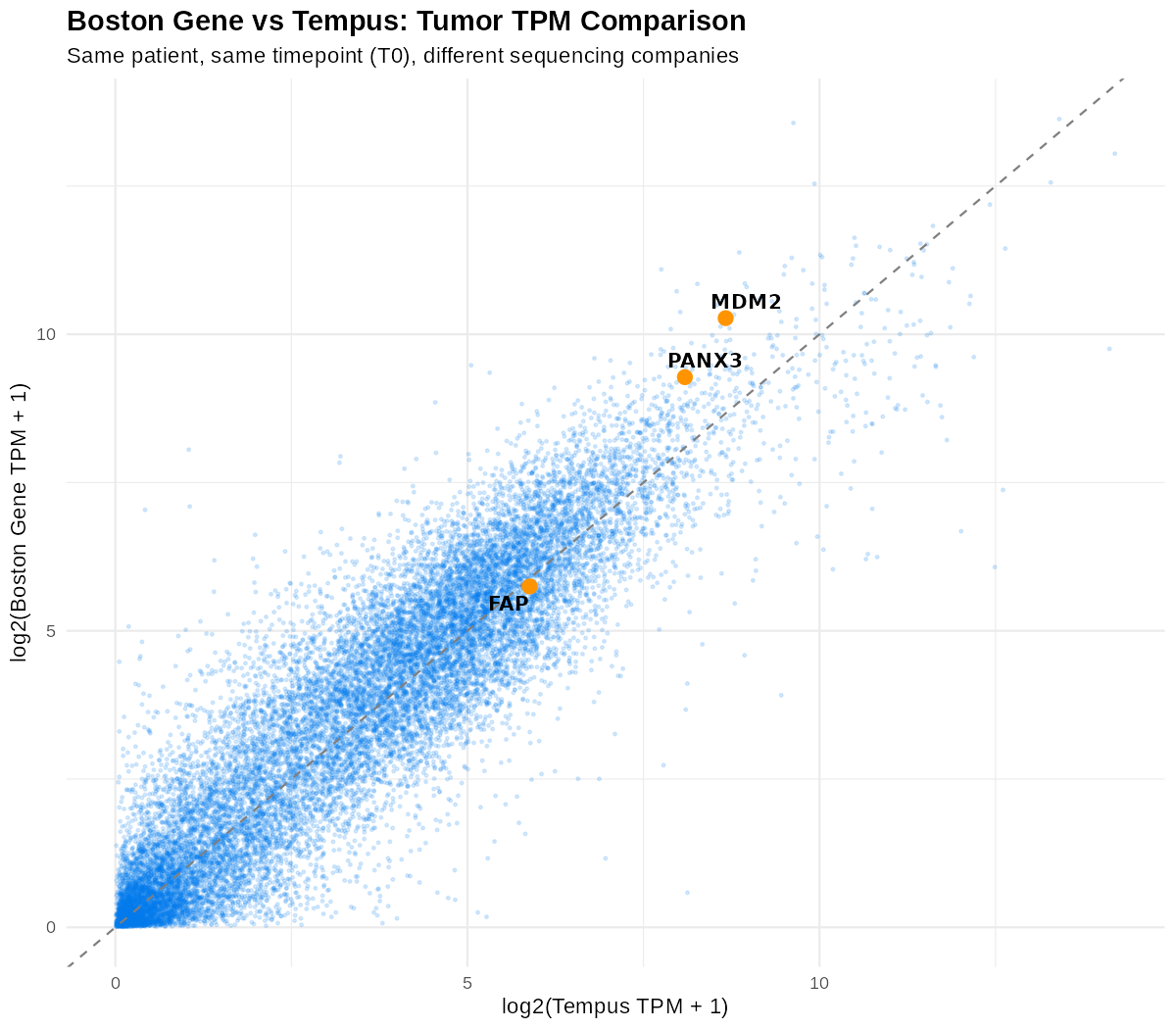
같은 환자·같은 시점(T0)을 두 회사에서 시퀀싱한 결과의 log2(TPM+1) 산점도. 완전한 직선은 아니지만 대각선을 따라 강한 상관을 보여, 발현량 정량이 재현성 있게 만들어졌음을 확인. 뒤에서 다룰 후보 유전자(MDM2·PANX3·FAP)의 위치도 표시돼 있다.
검증 ②: 이미 알려진 유전자가 실제로 튀는가
섹션 제목: “검증 ②: 이미 알려진 유전자가 실제로 튀는가”Sid 케이스에서 질병 관련 유전자로 명시됐던 것들이, 내 데이터에서도 같은 패턴으로 튀는지 봅니다.
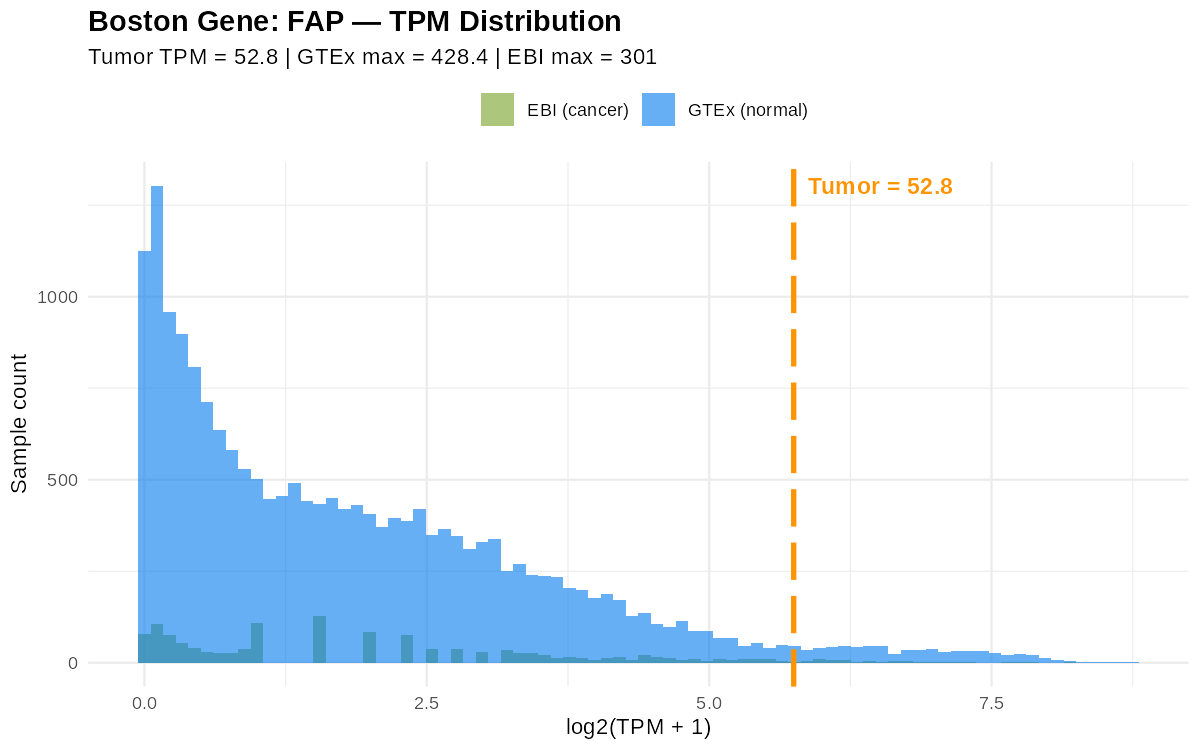
한 플롯이 유전자 하나에 대응한다. 가로축은 log2(TPM+1), 세로축은 샘플 수. 파란색은 GTEx(정상), 초록색은 EBI(암) 분포이고, 주황 점선이 내 종양 값이다. FAP는 원래 싱글셀 데이터에서 찾아낸 표적이라 total RNA에서는 신호가 아주 도드라지진 않지만(그래도 과발현 쪽), 주요 표적으로 거론된 다른 유전자들은 분포를 한참 벗어나 찍힌다.
읽는 법: 대부분의 샘플은 히스토그램 중심 근처에 발현량이 몰려 있고, 내 샘플(주황 선)이 그 중심에서 얼마나 오른쪽으로 벗어났는지가 곧 “튀는 정도”입니다. 이건 Sid가 공개 뷰어에서 보여준 그림과 같은 형식입니다.
7. 아웃라이어를 고르는 지표
섹션 제목: “7. 아웃라이어를 고르는 지표”유전자 4.5만 개를 눈으로 다 볼 수는 없으니, 수치로 아웃라이어를 추리는 지표가 필요합니다. 종양 TPM을 레퍼런스 분포와 견주는 네 가지를 씁니다.
| 지표 | 무엇을 보나 | 기준 예시 |
|---|---|---|
| 백분위 순위(percentile) | 내 TPM이 레퍼런스 분포에서 상위 몇 % 인가 | max · 상위 1% · 5% 등 |
| Z-score | 레퍼런스 평균에서 표준편차 몇 배 떨어졌나 | ∣Z∣ ≥ 2 |
| Fold change | 레퍼런스 중앙값(median)의 몇 배 인가 | log2FC ≥ 2 → 4배 이상 |
| Max comparison | 정상에서 관찰된 최댓값보다도 종양이 높은가 | True / False (직접 비교) |
-
Z-score가 특히 유용한 이유: 샘플이 하나뿐이라 p-value를 직접 구하긴 어렵습니다. 대신 레퍼런스 분포를 평균 0·표준편차 1로 표준화하고 내 값을 그 위에 찍으면, “분포에서 얼마나 극단인지”를 한 숫자로 표현할 수 있습니다.
-
Fold change는 로그2 스케일로 봅니다:
log2FC = 2 면 4배, = 9 면 512배입니다. (+1은 TPM이 0인 유전자에서 로그가 발산하지 않게 하는 보정.)
-
Max comparison은 논문에 자주 등장하는 정식 지표는 아니지만, Sid 케이스가 GTEx의 조직별 max TPM 과 직접 비교한 방식이라 재현 차원에서 넣었습니다. 수치가 아니라 True/False: “정상 최댓값조차 넘겼나?” 를 봅니다.
실제 계산: 알려진 표적 유전자들
섹션 제목: “실제 계산: 알려진 표적 유전자들”암(EBI/PCAWG) 레퍼런스로 계산한 결과의 일부입니다. ebi_median·ebi_max는 암 환자 약 1,350명의 그 유전자 발현량의 중앙값·최댓값입니다.
| gene | tumor TPM | ebi median | ebi max | z-score | log2FC | ≈ 배수 |
|---|---|---|---|---|---|---|
| PANX3 | 619.43 | 0.138 | 19.84 | 9.14 | ≈ 560× | |
| MDM2 | 1235.72 | 4.954 | 2731 | 4.47 | 5.32 | ≈ 40× |
| FAP | 52.76 | 2.0 | 301 | 1.97 | 3.75 | ≈ 13× |
PANX3를 보면: 암 환자 중앙값이 0.138인데 내 종양은 619.43. 백분위로 최댓값마저 넘고, z-score는 19.84(평균에서 표준편차 20배 거리!), 중앙값 대비 약 560배(log2FC 9.14) 발현입니다. 교과서적인 아웃라이어죠.MDM2는 Sid가 실제 표적으로 삼은 유전자로, 종양 TPM 1235.72: 공개 뷰어에서는 1500 언저리로 표시되는 값입니다.FAP는 신호가 상대적으로 약합니다(z ≈ 2). total RNA에서는 도드라지지 않는, 싱글셀에서 잡힌 표적이라는 점과 일치합니다.
1차 필터: 정상/암 최댓값 대비 2배 선
섹션 제목: “1차 필터: 정상/암 최댓값 대비 2배 선”여러 지표를 조합해 후보를 좁힙니다. 예컨대 종양 TPM을 EBI 암 max TPM과 직접 산점도로 놓고, 대각선(y=x)과 그 2배 선(log2 기준 +1)을 그으면:
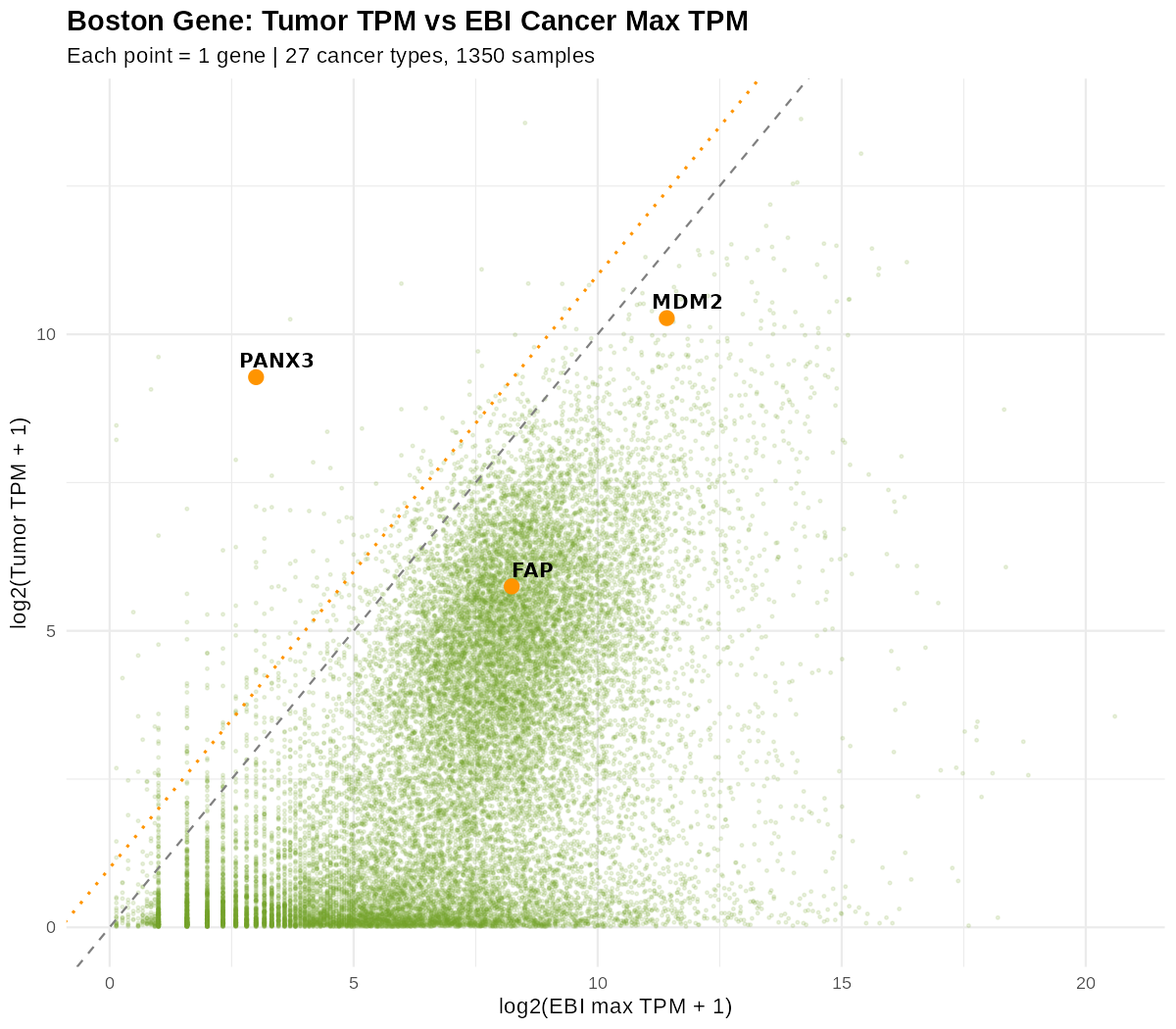
점 하나가 유전자 하나(27개 암종·1,350 샘플의 max 기준). 대각선(y=x)은 “암 최댓값과 같음”, 위쪽 점선은 그 2배 선. PANX3·MDM2처럼 2배 선을 넘어 위로 벗어난 유전자가 1차 아웃라이어 후보다. max 기준이라 종양에서만 튀는 완벽한 그림은 아니고(샘플 간 변이가 있어), 그래서 여러 지표를 함께 본다.
이렇게 레퍼런스 max·중앙값 대비 2배 이상 벗어난 유전자를 1차 아웃라이어로 추려 냅니다. 한 지표로 순위를 매길 수도, 여러 지표를 조합할 수도 있습니다.
8. 그다음: 기능 분석과 문헌조사
섹션 제목: “8. 그다음: 기능 분석과 문헌조사”아웃라이어 유전자 목록(예: 상위 100개)을 얻었으면, 이들이 어떤 생물학적 기능에 얽혀 있는지 봅니다.
g:Profiler / NetworkAnalyst 같은 기능 강화(enrichment) 분석 도구는, 유전자 리스트를 입력받아 “이 목록 안에 어떤 pathway·GO term에 속한 유전자가 많이 들어 있는지” 를 통계적으로(p-value와 함께) 알려 줍니다.
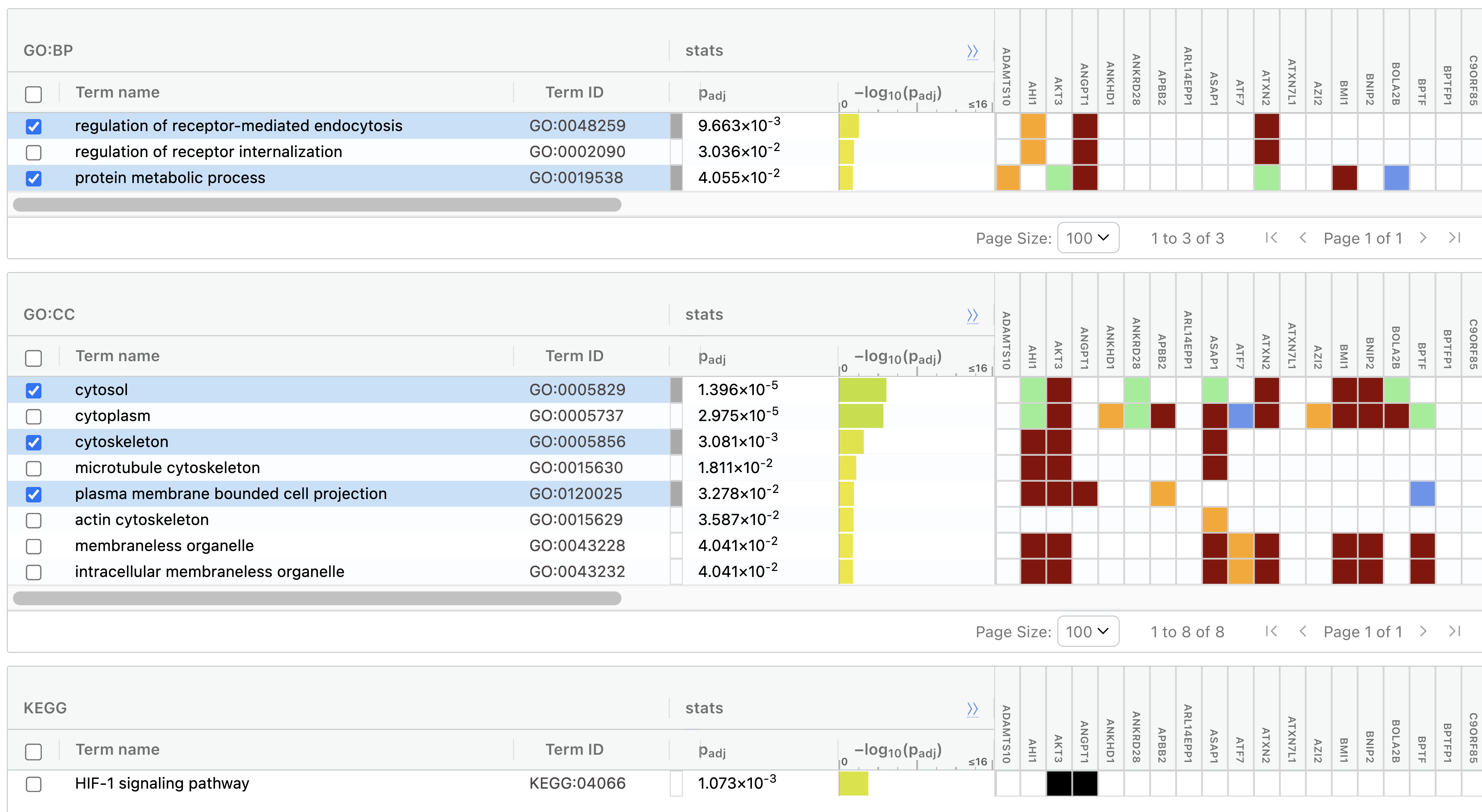
상위 100개 아웃라이어를 g:Profiler에 넣은 결과. HIF-1 신호(저산소 적응·악성화), cytoskeleton(세포 이동·전이), receptor-mediated endocytosis(신호 수용체 체내화·증폭) 등 종양과 맞닿은 term이 유의하게 떴다.
아쉬운 점도 솔직히: 골육종·암 term이 직접 뜨길 기대했지만 그렇게 선명하진 않았습니다. 그래도 이 유전자들이 주로 관여하는 pathway를 알면, 표적을 찾는 탐색 범위를 좁히는 나침반이 됩니다. 여기에 문헌조사(기존 약물·다른 질환에서의 표적 활용 여부)와 멀티오믹스(변이 탐색으로 과발현의 원인이 변이인지 확인 → 변이 표적 약물 설계)를 얹어 인과에 다가갑니다.
다음 시간 예고
섹션 제목: “다음 시간 예고”- HISAT2 + StringTie 조합 실습 보완 (STAR+RSEM의 대안 경로)
- 컨벤셔널 bulk RNA-seq 분석: 차등발현 유전자 분석(DEG, DESeq2), 그리고 GO / GSEA 기능 분석·시각화. (다른 public sample이 추가로 필요해 진행 여부는 논의)
- 못다 한 추가 실습은 각자 이어서 돌려 보시면 좋습니다.
오늘은 발현량 행렬을 실제로 만든 뒤, 그것으로 무엇을 하는지를 끝까지 따라갔습니다:
STAR 로그로 정렬 품질 확인 → RSEM(TPM)·featureCounts(count) 산출 → 한 샘플을 GTEx·PCAWG와 비교 → 백분위·z-score·fold change·max로 아웃라이어 추출 → 기능(pathway) 분석
핵심은, 샘플이 하나뿐이어도, 잘 고른 레퍼런스 분포 위에 내 값을 얹기만 하면, “비정상적으로 튀는 신호”를 정량적으로 집어낼 수 있다는 점입니다. Sid가 표적을 찾은 길이 바로 이것이었습니다. 다만 튄다 ≠ 원인이라는 것, 그래서 변이·pathway·문헌이 함께 가야 한다는 것을 잊지 않기로 합니다.
- 스터디 강의 자료 (
Day4: RNA analysis (2)) - 정렬 도구: STAR · Samtools
- 정량: RSEM · featureCounts (Subread)
- 정상 조직 발현 레퍼런스: GTEx Portal
- 암 발현 레퍼런스: PCAWG (Nature 2020) · EBI Expression Atlas
- 기능 강화 분석: g:Profiler · NetworkAnalyst
- Sid 공개 발현 뷰어: osteosarc.com/rnaseq