두 번째 수업: RNA-seq, FASTQ에서 발현량 행렬까지
첫 수업에서 우리는 Sid 데이터의 분자적 기원, DNA·RNA·단백질, 노말셀과 캔서셀, 그리고 오믹스 시퀀싱의 큰 그림, 을 따라갔습니다. 이번 수업은 그중 RNA-seq 한 갈래를 끝까지 파고듭니다: 조직에서 뽑은 RNA가 어떤 과정을 거쳐 시퀀서에 들어가고, 거기서 나온 원시 read가 어떤 단계를 거쳐 최종 발현량 행렬(expression matrix) 이 되는지.
오늘의 목표: RNA-seq 데이터가 생성되는 과정을 따라가며, FASTQ → 품질관리 → 트리밍 → 정렬 → 정량(TPM) 으로 이어지는 파이프라인 전체를 한 번에 꿴다. 그리고 Sid 사례에서 봤던 그림들이 바로 이 파이프라인을 통해 만들어졌음을 확인한다. 이번 시간은 시리즈의 RNA analysis (1): 데이터 생성 편이고, 만들어진 행렬을 해석하는 분석편은 다음 시간입니다.
1. 왜 RNA-seq인가 (복습)
섹션 제목: “1. 왜 RNA-seq인가 (복습)”한 몸의 모든 세포는 같은 DNA를 갖지만 켜는 유전자가 다릅니다. RNA-seq는 “지금 이 조직·세포가 어떤 유전자를 얼마나 켜고 있나(발현량)“를 측정합니다. 그래서 핵심 비교는 이겁니다:
정상 조직 vs. 변이(종양) 조직: 같은 유전자라도 발현량이 어떻게 다른가. 이 차이에서 어떤 메커니즘 변화가 어떤 표현형으로 이어지는지 단서를 얻습니다.
RNA-seq로 답할 수 있는 질문들:
-
변이 유전자가 실제로 발현되는지 확인: DNA에 변이가 있어도 그 유전자가 켜져 있지 않으면 단백질로 이어지지 않습니다.
-
표적·백신 후보 선정과 우선순위: 과발현 유전자·항원 후보를 추려 치료 타겟을 좁힘 (Sid의 개인화 치료로 직접 연결).
-
싱글셀(scRNA-seq)이라면 한발 더 나아가 어떤 세포가 그 유전자를 켜는지, 종양 미세환경·세포 간 상호작용까지 봅니다.
-
벌크(bulk) RNA-seq: 조직 전체를 갈아 섞인 평균 발현을 측정. 세포 유형별 발현은 구분되지 않으며, 주로 조직 수준의 발현 비교(DEG)에 활용.
-
싱글셀(single-cell) RNA-seq: 세포 하나하나를 따로 측정.
이번 수업은 Sid 케이스에서 실제로 쓰인 벌크 RNA-seq 파이프라인을 다룹니다.
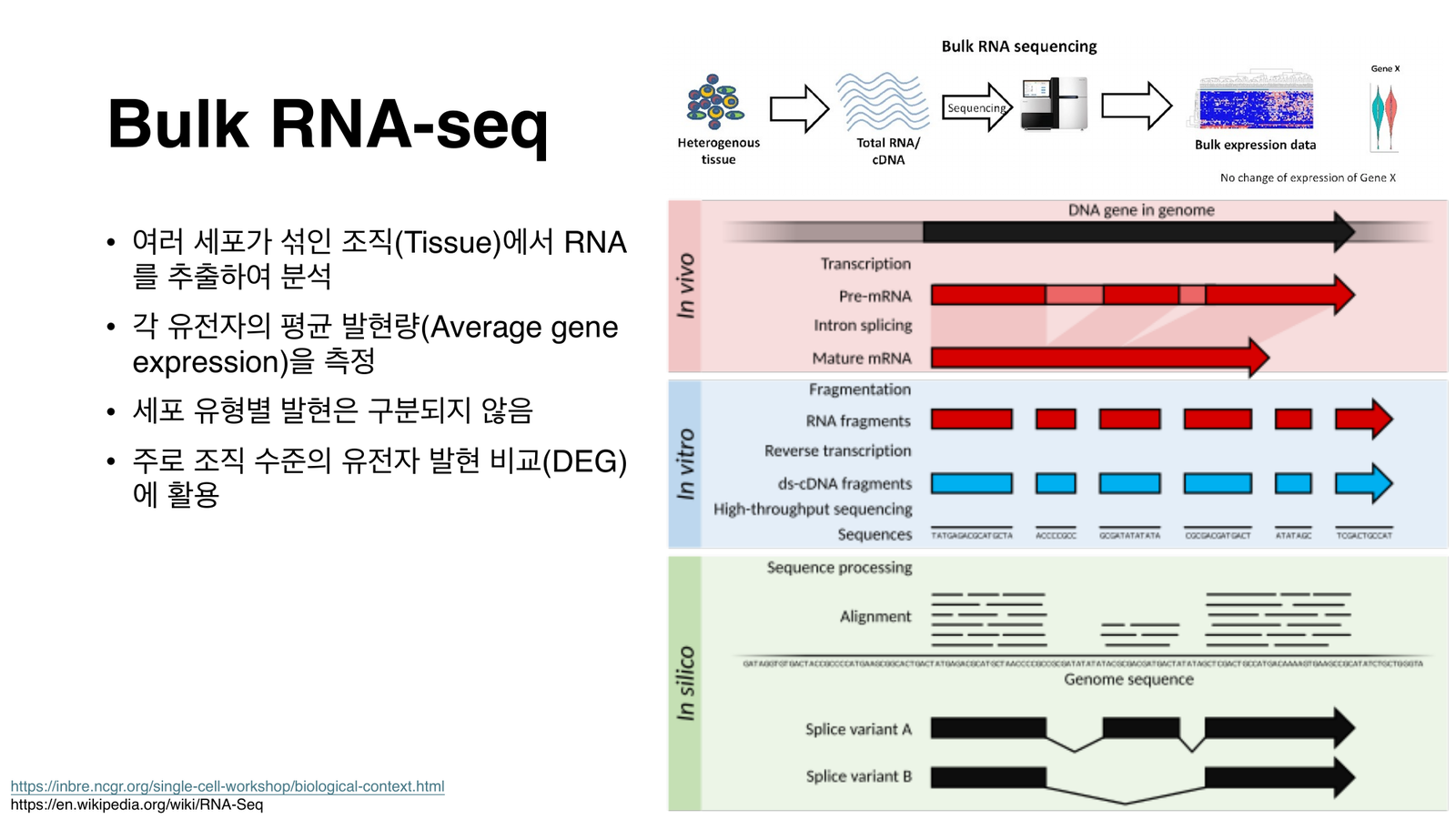
벌크 RNA-seq는 여러 세포가 섞인 조직에서 평균 발현량을 측정한다. 오른쪽은 유전자(DNA)가 전사·프로세싱을 거쳐 시퀀싱 데이터가 되고, 다시 게놈에 정렬되는 전 과정.
2. 시퀀싱 데이터는 어떻게 생성되나
섹션 제목: “2. 시퀀싱 데이터는 어떻게 생성되나”전체 흐름은 이렇습니다:
조직 샘플 → RNA 추출 → cDNA 합성 → 라이브러리 제작 → 시퀀싱 머신 → FASTQ read 생성
- 조직 샘플(tissue): 보통 한 조직의 일부를 떼어냅니다. 그 안에는 다양한 세포 타입이 섞여 있어 헤테로지니어스(heterogeneous) 합니다: 벌크가 “평균”을 보는 이유.
- RNA 추출 → cDNA: RNA는 단일 가닥이라 불안정하고, NGS는 DNA를 읽는 장비입니다. 그래서 mRNA에 상보 가닥을 붙여 안정적인 cDNA(complementary DNA) 로 합성한 뒤 시퀀싱합니다.
- 라이브러리 제작: cDNA를 잘게 조각내고 양 끝에 어댑터(adapter) 를 붙여, 시퀀서가 인식·증폭·읽을 수 있는 형태로 만듭니다.
🧩 in vivo / in vitro / in silico: in vivo = 생체 내에서, in vitro = 생체 밖(시험관)에서, in silico = 컴퓨터 상에서. 시료 채취·RNA 추출은 in vitro, read의 위치를 찾고 발현량을 세는 분석은 in silico입니다.
🧩 스플라이싱과 아이소폼(복습 연결): 첫 수업에서 봤듯, pre-mRNA에서 인트론이 잘려 나가고 엑손만 이어 붙어 성숙 mRNA가 됩니다. 그런데 어떤 엑손 조합이 쓰이느냐에 따라 같은 유전자에서 서로 다른 아이소폼(isoform) 이 나올 수 있습니다. 이 때문에 RNA-seq read를 게놈에 정렬할 때 특별한 처리가 필요합니다(→ 8번).
read는 mRNA보다 훨씬 짧다
섹션 제목: “read는 mRNA보다 훨씬 짧다”mRNA를 조각낸 뒤 시퀀싱하므로, 개별 read는 실제 mRNA보다 훨씬 짧습니다 (보통 100~150 bp).
- paired-end read: 하나의 fragment를 양쪽 끝에서 각각 읽는 방식. 두 read는 같은 read ID를 공유하며,
_1/_2두 파일로 나뉩니다.
🧩 fragment 길이 vs. read 길이: 시퀀서(예: Illumina)가 한 번에 읽을 수 있는 길이는 정해져 있습니다(예: ~100 bp). fragment가 200 bp면 양 끝을 읽어 거의 다 덮지만, 500 bp면 가운데는 읽히지 않고 양 끝 100 bp씩만 read로 나옵니다. 그래서 read 길이는 mRNA의 실제 길이가 아니라 라이브러리 fragment와 장비 사양이 정합니다.
paired-end가 한 fragment의 양 끝을 어떻게 읽는지 짧은 애니메이션으로 보면 직관적입니다 (Simple Science, 영어):
3. 시퀀서는 실제로 어떻게 염기를 읽나
섹션 제목: “3. 시퀀서는 실제로 어떻게 염기를 읽나”Illumina 계열은 합성하며 읽는(SBS, sequencing by synthesis) 방식입니다. fragment를 판 위에 고정하고, ACGT 염기를 하나씩 붙여가며 각 염기가 내는 형광 신호를 매 사이클 촬영합니다.
기계 안에서 실제로 어떻게 동작하는지는 Illumina 공식 애니메이션으로 보면 한결 명확합니다 (영어):
🧩 fragment가 붙어 신호를 내는 판의 물리 구조, flow cell · lane · cluster · tile, 는 플로우 셀과 레인 레퍼런스에서 그림으로 정리했습니다.
형광 강도 신호 → 베이스 콜링(base calling) → 신뢰도(Phred score) 산출
- 이 위치가 T라고 콜링됐다면 T 형광만 강하게 나온 것이고, A로 콜링됐지만 C 신호도 꽤 강했다면, A이긴 한데 애매, 품질 점수를 낮게 매깁니다.
- 여러 신호가 섞여 어느 하나도 확실히 튀지 않으면 염기를 결정하지 못하고
N으로 표기합니다.
🧩 시퀀싱 업체·플랫폼: 대부분의 단(short) read는 Illumina(NovaSeq/NextSeq 등)에서 나오고, 사실상 표준입니다. PacBio·Oxford Nanopore는 별도 포맷의 long read를 만듭니다. 어느 쪽이든 원시 read는 기본적으로 FASTQ 로 떨어집니다: 10x Genomics 싱글셀도 마찬가지. (수업 중 “쇼니드/페바이오” 등 구어 표기가 섞였는데, 정확히는 Illumina / PacBio / Oxford Nanopore입니다.)
4. 파일 포맷
섹션 제목: “4. 파일 포맷”RNA-seq에서 마주치는 핵심 포맷만 정리합니다. 형식 자체의 상세 표는 데이터 형식 레퍼런스에 따로 있습니다.
- FASTA (
.fa/.fasta): 염기 서열 또는 단백질 서열을 표현.>로 시작하는 description 라인 + 그 아래 서열 라인. 레퍼런스 게놈이 이 포맷입니다. - FASTQ (
.fq/.fastq): 시퀀서가 내놓는 원시 read. 4줄이 read 1개: ①@로 시작하는 ID, ② 서열, ③+(구분자), ④ 베이스별 품질(Phred→ASCII). 용량이 커서 보통.gz로 압축해 유통합니다. - BAM / SAM: 레퍼런스에 정렬된 결과. SAM은 사람이 읽는 텍스트, BAM은 그 압축 바이너리.
samtools로 다룹니다.
@SEQ_ID ← ① read IDGATTTGGGGTTCAAAGCAG ← ② 서열+ ← ③ 구분자!''*((((***+))%%%++ ← ④ 베이스별 품질 (서열과 길이 동일)
왼쪽은 실제 FASTA(>로 시작)·FASTQ 텍스트 예시, 오른쪽은 Phred 품질 점수와 오류 확률·정확도의 대응표.
5. 품질 관리: FastQC / MultiQC
섹션 제목: “5. 품질 관리: FastQC / MultiQC”read를 받으면 가장 먼저 분석할 만한 품질인지 확인합니다.
- FastQC: read 파일 하나를 읽어 품질 리포트를 생성. 보통 시퀀싱 직후와 트리밍 후 각각 수행합니다.
- MultiQC: 여러 샘플(paired-end이면 read마다)의 FastQC 리포트를 한 번에 모아 비교.
주요 확인 항목:
| 항목 | 기준 |
|---|---|
| Total Sequences | 20M~30M 이상 권장 (너무 적으면 샘플을 버리기도) |
| Per Base Sequence Quality | 대부분 Phred 30 이상 |
| Per Sequence Quality Score | 분포가 고품질 쪽에 집중 |
| Sequence Length Distribution | 트리밍 후 100 bp 언저리 |
| GC content | 샘플 내 read의 G+C 비율 |

FastQC가 보고하는 항목들. 파란색은 가급적 PASS가 필요한 지표, 빨간색은 RNA-seq에서 FAIL이 떠도 괜찮은 지표(Per base sequence content, Sequence duplication levels, Overrepresented sequences, Adapter content 등).
🔬 각 FastQC 항목이 실제로 무엇을 재고 그래프를 어떻게 읽는지: 항목별 상세는 QC(FastQC) 레퍼런스에 정리돼 있습니다.
6. 트리밍: Trim Galore! / Trimmomatic
섹션 제목: “6. 트리밍: Trim Galore! / Trimmomatic”저품질 구간과 어댑터 서열을 잘라내는 작업입니다.
- 주요 옵션: 말단 품질 기준(예: Q20 미만 제거), 최소 길이 미달 read 제거.
- 어댑터 서열은 라이브러리 종류에 따라 정해져 있어 툴이 대개 자동 인식합니다.
- 트리밍 후 다시 FastQC를 돌려, 저품질·어댑터가 제대로 제거됐는지 확인합니다.
✂️ 어댑터가 왜 생기고(read-through), polyG·polyA·N은 각각 무엇인지: fastp 기준 원리를 인터랙티브 위젯과 함께 트리밍 레퍼런스에서 볼 수 있습니다.
7. (확인) 트리밍 후 FastQC
섹션 제목: “7. (확인) 트리밍 후 FastQC”트리밍 결과가 크게 문제 없으면 정렬로 넘어갑니다. 이 시점의 read 길이가 예컨대 101 bp라면, 그건 mRNA 본래 길이가 아니라 라이브러리 fragment를 이 장비가 그만큼 읽은 결과입니다.
8. 정렬(alignment): STAR / HISAT2
섹션 제목: “8. 정렬(alignment): STAR / HISAT2”FASTQ read를 레퍼런스 게놈(FASTA) 과 유전자 주석(GTF) 에 맵핑해, 각 read가 게놈의 어디서 왔는지 찾습니다.
- RNA-seq의 특수성: junction read: 인트론이 제거된 mRNA에서 유래한 read는, 게놈 상에서 멀리 떨어진 두 엑손에 걸쳐 있을 수 있습니다. 이를 split 해서 정렬할 수 있는 splice-aware 정렬 도구가 필요합니다(STAR, HISAT2). 일반 DNA 정렬 도구로는 처리되지 않습니다.
- 인덱싱(indexing): 전체 게놈 서열을 미리 “위치 사전”으로 만들어 두는 작업. 정렬 속도를 크게 높입니다. 휴먼 레퍼런스는 미리 구축된 인덱스를 받아 쓸 수 있습니다.
- Sid 케이스에서는
STAR를 사용했습니다.
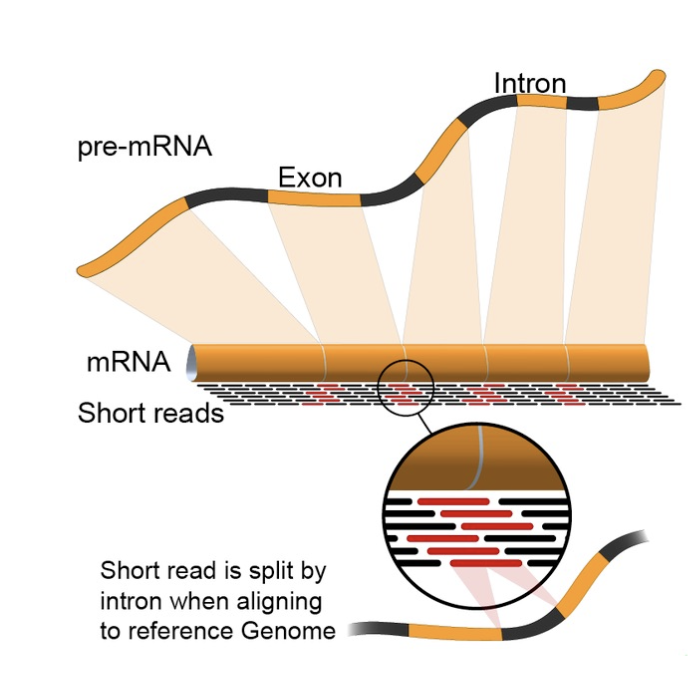
pre-mRNA에서 인트론이 제거돼 mRNA가 되고, 여기서 나온 short read를 게놈에 되돌려 정렬한다. 가운데 확대 부분처럼 한 read가 인트론을 사이에 둔 두 엑손에 걸쳐(splice) 나뉘어 정렬되기 때문에, 이를 처리하는 splice-aware 정렬 도구(STAR/HISAT2)가 필요하다.
9. 리드 카운트와 정규화: featureCounts / RSEM
섹션 제목: “9. 리드 카운트와 정규화: featureCounts / RSEM”정렬된 BAM에서 유전자마다 read 몇 개가 붙었는지 세고, 샘플 간 비교가 가능하도록 정규화합니다.
- 리드 카운팅 툴:
featureCounts,HTSeq,Salmon,RSEM등 → 유전자별·샘플별 read 수, 즉 raw count matrix 생성. - TPM (Transcripts Per Million): 유전자 길이와 시퀀싱 뎁스 두 가지를 모두 보정한 발현량 지표.
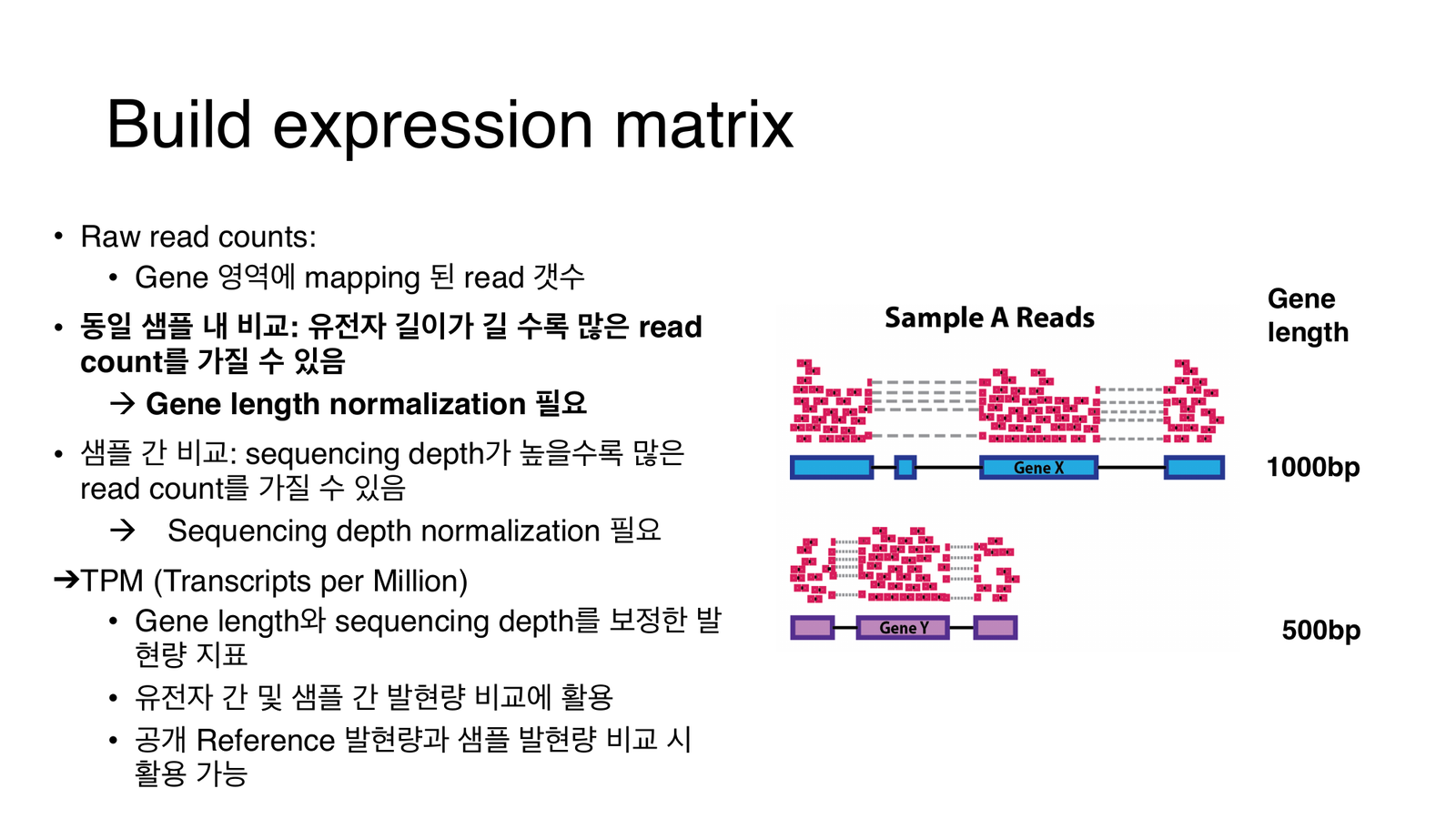
raw count는 두 가지에 휘둘린다: 유전자 길이(길수록 read가 더 붙음)와 시퀀싱 뎁스(많이 읽을수록 전체 count 증가). 둘 다 보정해야 유전자 간·샘플 간 비교가 가능하다.
TPM 계산 순서
- 유전자별 count를 유전자 길이로 나눠 RPK(reads per kilobase) 계산 → 긴 유전자일수록 read가 많이 붙는 편향을 보정.
- 한 샘플의 전체 RPK를 합산(= 시퀀싱 뎁스 추정)해서 그 값으로 나누고 ×10⁶.

작은 예제로 본 TPM 2단계 계산. Step 1에서 길이로 나눠 RPK를 구하고, Step 2에서 샘플의 전체 RPK 합으로 나눈 뒤 100만을 곱한다(슬라이드는 설명을 위해 ×10을 사용).
- TPM은 같은 샘플 내 유전자 간 비교와 샘플 간 비교 모두에 쓸 수 있습니다.
- RSEM: 정렬부터 TPM 계산까지 포함해 결과를 내며, Sid 케이스에서 실제로 TPM을 만든 도구입니다.
🎛️ 왜 정규화가 필요하고 RPKM/FPKM과 TPM이 어떻게 다른지: 슬라이더로 직접 만져 보며 이해하려면 TPM와 정규화 심화 문서로.
💡 한 줄 요약: raw count(featureCounts) 는 “몇 개 붙었나”, TPM(RSEM) 은 “길이·뎁스를 보정해 서로 비교 가능하게 만든 값”. 최종 발현량 행렬이 이렇게 완성됩니다.
10. Sid 케이스에 어떻게 쓰였나
섹션 제목: “10. Sid 케이스에 어떻게 쓰였나”이 파이프라인의 산출물(TPM 행렬)을, Sid 팀은 두 공개 레퍼런스와 나란히 비교해 “본인에게서만 이상하게 튀는 유전자”를 찾았습니다.
| 레퍼런스 | 규모 | 쓰임 |
|---|---|---|
| GTEx | 약 950명 · 54개 조직 · 약 2만 개 샘플 | 정상 조직 대비 발현량 비교 (조직별 max TPM 기준) |
| PCAWG | 2,658명 · 38개 암종 | 다른 암 환자들과 비교해 이상 발현 확인 |
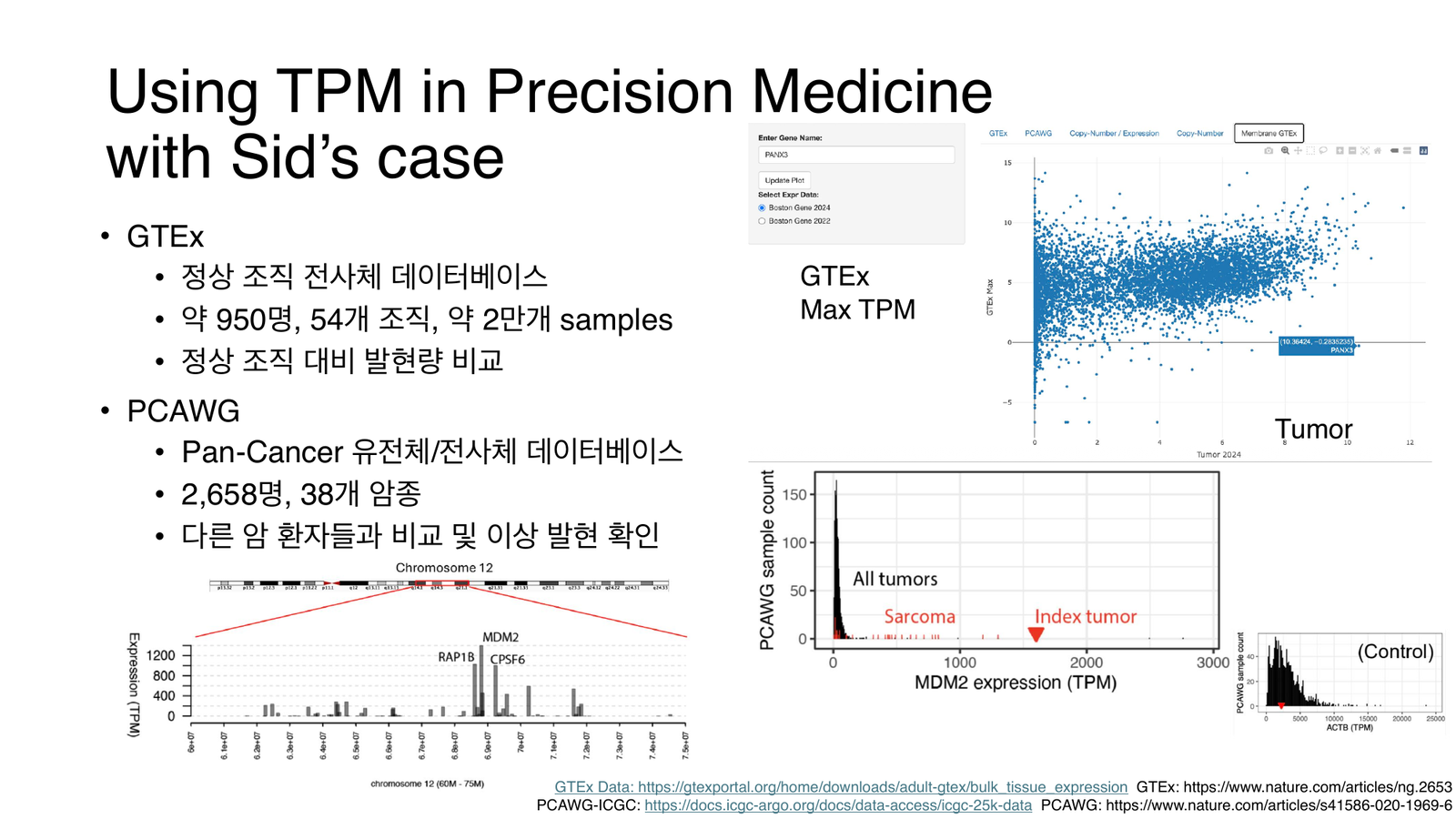
왼쪽 산점도: GTEx 정상 조직의 조직별 max TPM(x축) 대비 본인 종양 TPM(y축): 정상에서 낮은데 본인에게서 튀는 점이 후보다. 오른쪽 히스토그램: PCAWG 암 환자 분포 안에서 본인 종양(index tumor)·육종(sarcoma)이 어디 위치하는지.
비교는 Sid가 공개한 인터랙티브 뷰어 osteosarc.com/rnaseq에서 직접 볼 수 있고, 유전자 하나에 대해 세 가지 화면을 제공합니다:
- 유전자별 발현(Genes): 염색체상 유전자별 TPM을 시각화해 특이적으로 높은 유전자 탐색.
- 레퍼런스 비교(Assay Comparison): GTEx(정상) · PCAWG(암) 대비 본인 종양 TPM이 어디에 위치하는지 산점도·히스토그램으로 확인.
- 복제수 연계(Copy Number): copy number variant(CNV) 와 발현량을 함께 봐, 유전자 증폭이 과발현으로 이어졌는지 확인.
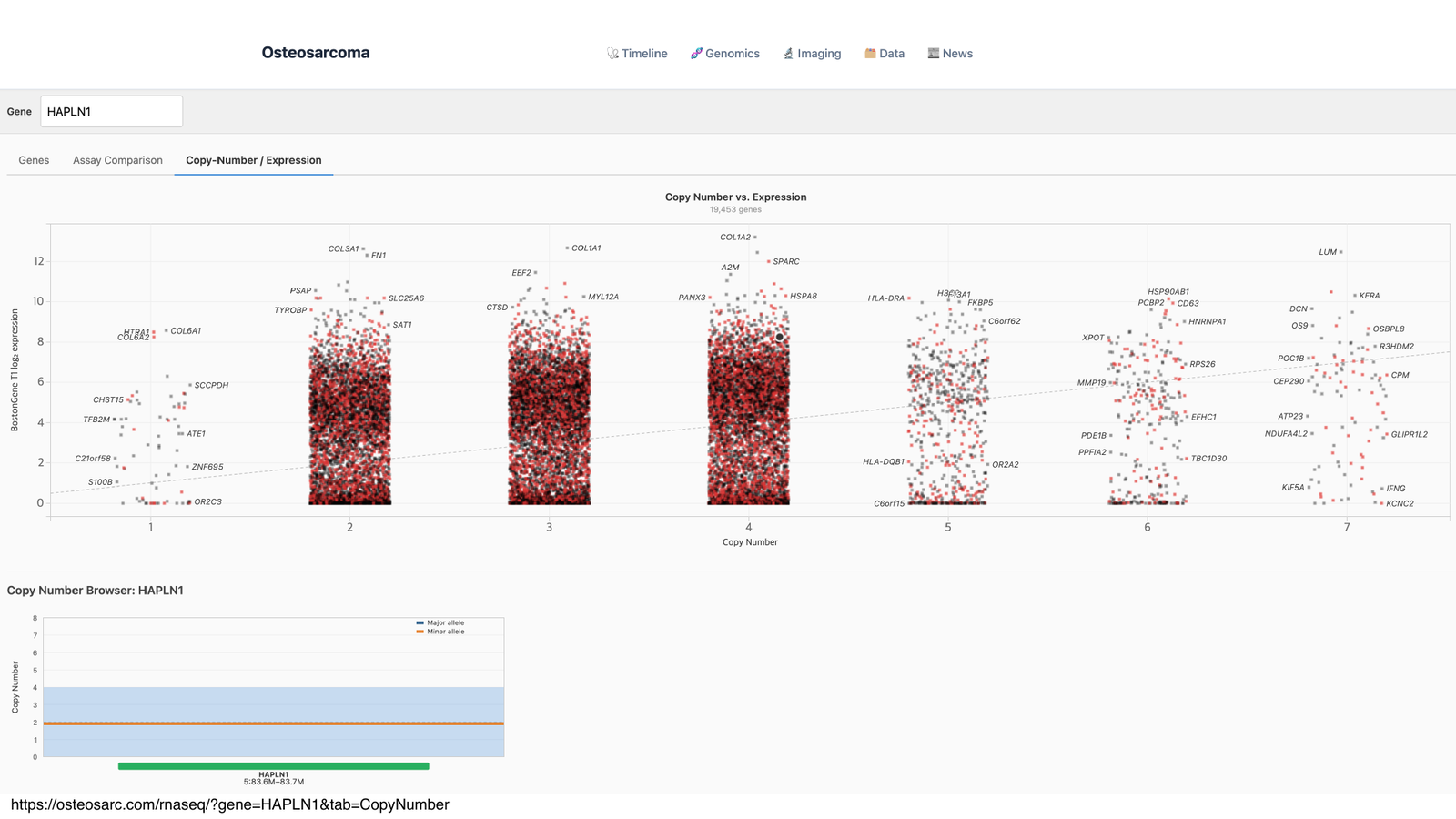
뷰어의 Copy-Number / Expression 탭. 위는 유전자 복제수(x축)와 발현량(y축)의 관계: 복제수가 늘수록 발현이 오르는 경향을 보고, 아래는 선택 유전자(HAPLN1)의 복제수 브라우저.
💡 수업에서는
HAPLN1을 예시로 뷰어를 둘러봤습니다. 그리고 바로 이 흐름으로 Sid의 ⑤ 표적치료 근거가 된 MDM2 과발현이 발견됐습니다: 정상·타 환자 대비 비정상적으로 높은 한 줄의 신호가 표적이 된 것이죠. 또한 여러 시점(timepoint) 의 TPM 변화를 비교해 치료 전후 발현 추이도 추적했습니다.
🧩 CNV(유전자 복제수 변이)와 발현량을 함께 보는 분석은, 추후 DNA 전장유전체(WGS) 분석 파트에서 더 깊이 다룰 예정입니다.
11. 실습: 공개·실제 데이터로 끝까지 돌려보기
섹션 제목: “11. 실습: 공개·실제 데이터로 끝까지 돌려보기”수업에서 받은 Day3 Practice 자료의 핸즈온 구성입니다. 목표는 FastQC → 트리밍 → STAR 정렬 → featureCounts(raw count) + RSEM(TPM) → GTEx 비교 까지 직접 한 번 돌려보는 것.
워크플로우
섹션 제목: “워크플로우”- FastQC: raw read 품질 확인
- Trim Galore!: 트리밍(저품질·어댑터 제거)
- FastQC: 트리밍된 read 재확인 (결과가 크게 문제없으면 다음 단계로)
- STAR: 리드 정렬 (미리 만들어진 index 사용 가능)
- featureCounts: raw read count 계산
- RSEM: TPM 계산
단계별 핵심 명령
섹션 제목: “단계별 핵심 명령”수업 슬라이드에 나온 예시 명령(파일명·경로는 자리표시자):
# 1) raw read QCfastqc sample_R1.fastq.gz sample_R2.fastq.gz
# 2) 트리밍 (말단 Q20 미만 / 길이 20bp 미만 제거)trim_galore --paired --quality 20 --length 20 sample_R1.fastq.gz sample_R2.fastq.gz# → sample_R1_val_1.fq.gz / sample_R2_val_2.fq.gz
# 3) 트리밍된 read 재QCfastqc sample_R1_val_1.fq.gz sample_R2_val_2.fq.gz
# 4) STAR 인덱싱 (미리 만들어진 인덱스가 있으면 생략)STAR --runThreadN 6 --runMode genomeGenerate --genomeDir ./ref \ --genomeFastaFiles ref.fa --sjdbGTFfile ref_genes.gtf
# 5) STAR 정렬 → 좌표 정렬된 BAMSTAR --genomeDir ./ref --readFilesIn sample_R1_val_1.fq.gz sample_R2_val_2.fq.gz \ --runThreadN 8 --outSAMtype BAM SortedByCoordinate
# 6) 유전자별 raw read count (여러 BAM을 한 번에도 가능)featureCounts -a ref_genes.gtf -o featureCounts_output.txt sample_sorted.bam
# 7) TPM 계산은 RSEM 사용 (Sid 케이스에서 실제로 쓰인 도구)데이터 구성
섹션 제목: “데이터 구성”| 구분 | 내용 |
|---|---|
| 샘플 | T0 종양(tumor) 샘플: Tempus(약 1.2 GB / 뎁스 14.2M), BostonGene(약 10 GB / 뎁스 74.3M). paired-end이라 R1/R2 두 파일 |
| 레퍼런스 게놈 | GRCh38 (정렬에는 GRCh38_noALT_noHLA_noDecoy STAR 인덱스 사용) |
| 유전자 주석 | GENCODE v47 GTF |
| 공개 비교 데이터 | GTEx (정상 조직 gene TPM), PCAWG (암 발현 레퍼런스) |
사용 도구
섹션 제목: “사용 도구”| 단계 | 도구 |
|---|---|
| Read QC | FastQC |
| Trimming | Trim Galore! |
| Alignment | STAR |
| SAM/BAM 처리 | Samtools |
| Raw count | featureCounts (Subread) |
| TPM | RSEM: Sid 케이스 사용 |
자주 쓰는 명령 (Linux 기준)
섹션 제목: “자주 쓰는 명령 (Linux 기준)”# 링크로 데이터 다운로드wget [link]
# .tar.gz 압축 해제tar -zxvf [파일이름].tar.gz
# .gz 압축 해제gzip -d [파일이름].gz
# .gz 파일 미리보기 (압축 풀지 않고 앞부분만)zcat [파일이름].gz | head
# BAM 파일을 사람이 읽는 SAM 형태로 보기samtools view [파일이름].bam | head다음 주 계획: Expression matrix 분석
섹션 제목: “다음 주 계획: Expression matrix 분석”- Sid 케이스 따라 실습: 실제 read에서 TPM 직접 계산 → GTEx·PCAWG와 비교 → candidate gene 탐색·기능 분석
- (옵션) 컨벤셔널 bulk RNA-seq 분석: 차등발현(DEG, DESeq2), GO / GSEA 기능 분석: 다른 public sample이 필요해 진행 여부는 논의
- 추가 도구·환경: IGV(정렬 BAM 시각화), R(≥4.3) · Bioconductor(≥3.18) · RStudio
오늘 우리는 Sid 데이터의 RNA-seq 파이프라인을 처음부터 끝까지 따라갔습니다:
조직 → cDNA → 라이브러리 → 시퀀싱(FASTQ) → FastQC → 트리밍 → STAR 정렬 → featureCounts(count) / RSEM(TPM) → 발현량 행렬 → GTEx 비교
Sid 사례에서 봤던 “정상 대비 튀는 유전자” 그림은 마법이 아니라, 이 단계들을 차례로 통과한 결과였습니다. 다음 수업에서는 이 길을 직접 손으로 한 번 걸어봅니다.
- 스터디 강의 자료 (
Day3: RNA analysis (1)) 및 실습 가이드(Day3 Practice) - 레퍼런스 게놈 ALT/HLA/decoy 제거 방식: GTEx / TOPMed RNA-seq pipeline
- 정상 조직 발현 레퍼런스(GTEx): GTEx Portal: bulk tissue expression · Nature Genetics 2013
- 암 발현·유전체 레퍼런스(PCAWG): Pan-cancer analysis of whole genomes, Nature 2020 · ICGC/ARGO 데이터 접근
- Sid 공개 발현 뷰어: osteosarc.com/rnaseq