QC: read 품질 검사
정렬에 들어가기 전에 받은 read가 쓸 만한지, 어디를 손봐야 하는지 확인하는 첫 단계입니다. 파이프라인에서의 위치:
FASTQ 확보 → [① QC (FastQC)] → ② 트리밍 → ③ 정렬 → ④ 정량(TPM) → ⑤ 비교FastQC 소프트웨어 소개
섹션 제목: “FastQC 소프트웨어 소개”FastQC는 시퀀싱 read의 품질을 진단하는, 바이오인포매틱스에서 가장 널리 쓰이는 QC 도구입니다.
| 항목 | 내용 |
|---|---|
| 개발 | 영국 Babraham Institute(Babraham Bioinformatics), 주 저자 Simon Andrews |
| 라이선스 | GPL v3: 오픈소스, 자유 사용·수정·재배포 |
| 언어 | Java (크로스플랫폼: macOS·Linux·Windows) |
| 실행 방식 | GUI(클릭) 또는 CLI(명령줄) 둘 다 지원 |
| 입력 | FASTQ(여러 인코딩), 그리고 SAM/BAM(정렬 결과)도 가능 |
| 소스 | github.com/s-andrews/FastQC · 배포 페이지 |
오픈소스 운영·저장소
섹션 제목: “오픈소스 운영·저장소”- 저장소(소스): github.com/s-andrews/FastQC: GPL v3(번들된 JHDF5 라이브러리는 Apache-2.0). 컴파일된 실행 파일(Windows·macOS·Linux)은 Babraham 배포 페이지에서, 소스는 GitHub에서 받습니다. Bioconda·리눅스 배포판 패키지로도 널리 배포됩니다(우리가 설치한 것도 이 경로).
- 운영 형태: 대형 재단이 아니라 학술 기관(Babraham)의 코어 시설 도구로, 사실상 단일 메인테이너(Simon Andrews)가 이끄는 소규모 프로젝트입니다. 총 커밋 ~193개, 최신 릴리스 v0.12.1 (2023-03), 열린 이슈·PR 수십 개 수준으로 완만하지만 유지되는 활동입니다. 기여는 GitHub 이슈/PR로 받습니다.
- 즉 “거대 커뮤니티가 굴리는” 프로젝트라기보다, 한 연구소가 만들어 무료 공개하고 커뮤니티가 표준으로 채택한 형태입니다. (우리가 쓴 버전도 v0.12.1)
기술 스택
섹션 제목: “기술 스택”| 구성 | 내용 |
|---|---|
| 주 언어 | Java 81.6% (핵심 로직·GUI). 리포트 템플릿 HTML 16.1%, 실행 래퍼 Perl 1%·Python 1%·Shell 0.2%, 리포트 변환 XSLT 0.1% |
| GUI | Java Swing (클릭 방식 데스크톱 앱) |
| 빌드 | Apache Ant (build.xml) |
| 핵심 라이브러리 | HTSJDK (htsjdk.jar: SAM/BAM 읽기), Apache Commons Compress/IO (gzip·bzip2 등 압축 입력), JHDF5 (cisd-jhdf5.jar: 나노포어 FAST5의 HDF5 읽기) |
- Java라서 JVM만 있으면 어디서든 동일하게 돕니다(크로스플랫폼). HTSJDK를 품고 있어 FASTQ뿐 아니라 BAM도 직접 열 수 있습니다.
어떻게 동작하나
섹션 제목: “어떻게 동작하나”- 파일을 한 번 스트리밍으로 훑으며 여러 통계를 동시에 집계합니다. 레퍼런스 게놈이 필요 없고, 계산이 가벼워 몇 분이면 끝납니다.
- 데이터를 만들지도 바꾸지도 않습니다. FASTQ 4번째 줄의 품질 점수는 시퀀서가 판독하며 매긴 값이고, FastQC는 그걸 읽어서 요약할 뿐입니다.
- 중복도·과다서열처럼 전체를 다 기억할 수 없는 항목은 처음 등장하는 서열을 표본으로 추정합니다(메모리 절약).
FastQC가 읽는 것: FASTQ 한 read 뜯어보기
섹션 제목: “FastQC가 읽는 것: FASTQ 한 read 뜯어보기”FastQC가 요약하는 원재료는 FASTQ의 read 하나 = 4줄입니다. 실제 read 예시:
@DRR024878.1 HWI-ST1290:260:C5GAPACXX:4:1101:2723:1997/1 ← ① ID·장비 좌표NGTCAGGTGGTCACCCATCTTCTTGATAAGCTTCACTTCCTCATCTAGGAAG... ← ② 염기 서열+ ← ③ 구분자#1:DDFFEHHDHHIJJJJJJJJJJHIJJJJJJJEIIJJJJJJIJJJJFJHHJG... ← ④ 품질 점수- ① ID 줄(
@로 시작): read 번호 + 장비·flow cell·lane·tile·좌표. (좌표 의미는 Flow cell · Lane) - ② 염기 서열: 실제 읽은 A/T/G/C. 맨 앞
N은 판독 실패(어느 염기인지 결정 못 함). - ③ 구분자: 서열 끝·품질 시작 표시.
- ④ 품질 점수: ②와 길이가 같고, 각 염기가 얼마나 믿을 만한지를 문자 하나로 나타냄.
품질 점수(Phred)는 무슨 뜻인가
섹션 제목: “품질 점수(Phred)는 무슨 뜻인가”④의 값은 Phred quality score(Q): 그 염기가 틀렸을 확률을 로그로 나타낸 것입니다. 시퀀서가 판독하며(base calling) 직접 매겨 파일에 써넣은 값이고, FastQC는 이걸 만들지 않고 읽어서 요약만 합니다.
Q = -10 × log₁₀(P) (P = 판독 오류 확률)| Q값 | 오류 확률 | 정확도 |
|---|---|---|
| 10 | 1/10 | 90% |
| 20 | 1/100 | 99% (최소 기준선) |
| 30 | 1/1,000 | 99.9% (흔한 합격선) |
| 40 | 1/10,000 | 99.99% |
문자로 어떻게 저장되나: Phred+33
섹션 제목: “문자로 어떻게 저장되나: Phred+33”Q값(2, 16, 41…)을 염기 1개 = 문자 1개로 맞추려고 ASCII 문자로 인코딩합니다(Phred+33 방식).
문자 = ASCII( Q + 33 ) ⇄ Q = ASCII값(문자) − 33예를 들어 위 read의 앞부분을 풀면:
| 위치 | 염기 | 품질문자 | Q값 | 해석 |
|---|---|---|---|---|
| 1 | N | # | 2 | 판독 실패 → 그래서 염기가 N |
| 2 | G | 1 | 16 | 아직 낮음 |
| 3 | T | : | 25 | 올라오는 중 |
| 4 | C | D | 35 | 좋음 |
→ 맨 앞 1~2 염기는 품질이 낮다가 곧 Q35+로 안정되는 전형적 패턴(첫 사이클 신호 불안정). FastQC의 Per base sequence quality 그래프는 이런 위치별 Q값을 모든 read에 대해 모아 상자그림으로 그린 것입니다.
⚠️ 오래된 Illumina 데이터는 Phred+64를 쓰기도 했습니다. 요즘 데이터는 대부분 Phred+33(FastQC의 Basic Statistics에
Sanger / Illumina 1.9로 표시)이고, 도구가 자동 감지합니다.
산출물
섹션 제목: “산출물”파일당 두 가지가 나옵니다.
*_fastqc.html: 사람이 보는 리포트(그래프 + 신호등).*_fastqc.zip: 기계가 읽는 원자료:summary.txt(항목별 pass/warn/fail),fastqc_data.txt(수치), 그래프 이미지. → 자동화·MultiQC 취합에 사용.
신호등(pass/warn/fail)의 의미
섹션 제목: “신호등(pass/warn/fail)의 의미”각 항목마다 “이 값이면 경고/실패”라는 임계값이 내장돼 있습니다(아래 상세). 단 이 기준은 genomic DNA를 가정한 것이라, RNA-seq에선 정상인데 빨강이 뜨기도 합니다(뒤의 ⚠️ 참고). 임계값은 limits.txt로 조정할 수도 있습니다.
왜 QC를 먼저 하나
섹션 제목: “왜 QC를 먼저 하나”Garbage in, garbage out. 품질 나쁜 read를 그대로 정렬하면 잘못 붙거나 안 붙어서 뒤 결과가 다 흔들립니다. QC의 목적은 두 가지입니다.
- 데이터 진단: 시퀀싱이 잘 됐나, 이상은 없나
- 다음 단계(트리밍) 근거 마련: 어댑터가 있나? read 끝 품질이 떨어지나? → 무엇을 얼마나 자를지 결정
FASTQ 4번째 줄의 품질 점수(Phred)를 read 전체에 대해 그래프로 요약해 보는 것이 QC입니다. FASTQ 형식과 품질 점수는 데이터 형식 레퍼런스를 참고하세요.
도구: FastQC
섹션 제목: “도구: FastQC”가장 표준적인 QC 도구입니다. read 파일을 넣으면 HTML 리포트를 만들어, 여러 항목(module)을 초록(pass)/노랑(warn)/빨강(fail) 신호등으로 보여줍니다.
fastqc sample_1.fastq.gz sample_2.fastq.gz# → sample_1_fastqc.html, sample_2_fastqc.html 생성.gz를 그대로 읽습니다(압축 해제 불필요).- FastQC는 데이터를 바꾸지 않습니다. 원본 FASTQ는 그대로 두고, 진단 리포트만 새로 만듭니다. (실제로 자르는 것은 다음 단계 트리밍)
- paired-end 데이터는
_1,_2를 각각 검사합니다(FastQC는 짝 관계를 모름 → 리포트도 2개).
핵심 항목 읽는 법
섹션 제목: “핵심 항목 읽는 법”무엇을 보는지 네 가지 질문으로 묶으면 이해가 쉽습니다.
① “각 염기를 믿을 수 있나?”: 품질
섹션 제목: “① “각 염기를 믿을 수 있나?”: 품질”- Per base sequence quality: 위치별 품질. read 끝으로 갈수록 떨어지는지가 핵심 (→ 끝 트리밍 판단)
- Per sequence quality scores: read별 평균 품질. 대부분 Q30↑에 몰리면 좋음
- Per base N content: 판독 실패(N) 비율. 특정 위치에 몰리면 장비 이상
② “섞인 게 없나?”: 오염·인공물
섹션 제목: “② “섞인 게 없나?”: 오염·인공물”- Adapter content: 어댑터 서열 함량 (→ 올라가면 어댑터 트리밍)
- Overrepresented sequences: 과다 등장 서열 (어댑터·rRNA 오염 단서)
③ “서열 조성이 정상인가?”: 조성
섹션 제목: “③ “서열 조성이 정상인가?”: 조성”- Per base sequence content: 위치별 A/T/G/C 비율
- Per sequence GC content: GC 함량 분포 (이상한 봉우리 = 오염 의심)
④ “구조는 정상인가?”: 분포
섹션 제목: “④ “구조는 정상인가?”: 분포”- Sequence length distribution: read 길이 분포
- Sequence duplication levels: 같은 read 중복도
실습에서 실제로 행동을 바꾸는 건 대개 Per base quality(끝 품질 떨어지나?)와 Adapter content(어댑터 있나?) 둘입니다.
항목별 상세
섹션 제목: “항목별 상세”FastQC v0.12 기준 주요 모듈입니다. 각 모듈이 무엇을 재고, 어떤 값에서 경고(WARN)·실패(FAIL)를 띄우는지 정리합니다. (임계값은 대략적 기본값이며 limits.txt로 조정 가능)
Basic Statistics
섹션 제목: “Basic Statistics”파일 요약: 총 read 수, read 길이, GC%, 인코딩(예: Sanger/Illumina 1.9 = Phred+33). 항상 PASS에 가깝고, 인코딩·길이를 확인하는 용도.
Per base sequence quality
섹션 제목: “Per base sequence quality”각 위치(cycle)별 품질 점수 분포를 상자그림으로. 시퀀싱 특성상 뒤로 갈수록 낮아지는 게 정상.
- WARN: 어느 위치든 하위 사분위 <10 또는 중앙값 <25 · FAIL: 하위 사분위 <5 또는 중앙값 <20
- 끝부분이 크게 낮으면 → 끝 quality 트리밍의 근거.
Per tile sequence quality
섹션 제목: “Per tile sequence quality”flow cell의 tile(구획)별 품질 편차. 특정 tile만 나쁘면 그 구획에 기포·먼지 등 물리적 문제가 있었다는 뜻. (flow cell·tile 개념은 Flow cell · Lane)
- WARN: 어떤 tile이 해당 위치 평균보다 2 이상 낮음 · FAIL: 5 이상 낮음
Per sequence quality scores
섹션 제목: “Per sequence quality scores”read별 평균 품질의 분포. 대부분의 read가 높은 Q(≥27~30)에 몰려야 좋음. 낮은 쪽에 봉우리가 있으면 일부 read가 전반적으로 부실.
- WARN: 최빈 평균 품질 <27 (오류 0.2%) · FAIL: <20 (오류 1%)
Per base sequence content
섹션 제목: “Per base sequence content”위치별 A/T/G/C 비율. 이론상 위치와 무관하게 평평해야 함.
- WARN: 어느 위치서 A–T 또는 G–C 차이 >10% · FAIL: >20%
- ⚠️ RNA-seq은 앞 10여 bp가 치우쳐 거의 항상 FAIL: 정상(아래 참고).
Per sequence GC content
섹션 제목: “Per sequence GC content”read별 GC% 분포가 종 특유의 정규분포를 따르는지. 예상과 다른 봉우리는 오염(다른 생물·어댑터·rRNA) 신호일 수 있음.
- WARN: 이론 분포에서 벗어난 read가 >15% · FAIL: >30%
Per base N content
섹션 제목: “Per base N content”위치별 N(판독 실패) 비율. 정상은 0에 가까움. 특정 위치에서 치솟으면 장비/화학 문제.
- WARN: 어느 위치든 N >5% · FAIL: >20%
Sequence Length Distribution
섹션 제목: “Sequence Length Distribution”read 길이 분포. 트리밍 전이면 대개 균일(예: 100 bp).
- WARN: 길이가 제각각 · FAIL: 길이 0인 read 존재
- ⚠️ 트리밍 후엔 길이가 다양해져 WARN이 뜨는 게 정상.
Sequence Duplication Levels
섹션 제목: “Sequence Duplication Levels”같은 서열이 몇 번 중복되는지. 처음 등장 서열을 표본으로 추정.
- WARN: 비고유(중복) read >20% · FAIL: >50%
- ⚠️ RNA-seq은 고발현 유전자가 같은 read를 대량 생산 → 중복 높음이 정상.
Overrepresented sequences
섹션 제목: “Overrepresented sequences”전체의 일정 비율 이상 차지하는 특정 서열 목록 + 추정 출처. 어댑터·rRNA·polyA 꼬리 등이 잡힘.
- WARN: 어떤 서열이 >0.1% · FAIL: >1%
Adapter Content
섹션 제목: “Adapter Content”위치별로 알려진 어댑터 서열(Illumina Universal, Nextera, small RNA, polyA/G 등) 함량이 얼마나 누적되는지.
- WARN: 어떤 어댑터가 어느 위치서 >5% · FAIL: >10%
- 올라가면 → 어댑터 트리밍의 직접 근거.
참고: 예전 버전의 Kmer Content 모듈은 v0.11부터 기본 비활성화됐습니다.
⚠️ RNA-seq에서 FAIL이 떠도 정상인 경우
섹션 제목: “⚠️ RNA-seq에서 FAIL이 떠도 정상인 경우”FastQC는 원래 genomic DNA를 가정하고 만든 도구라, RNA-seq에선 정상인데도 빨강(fail) 이 뜨는 항목이 있습니다. 이걸 모르면 멀쩡한 데이터를 과하게 손대게 됩니다.
- Per base sequence content: 앞 10~13 bp가 들쭉날쭉: RNA-seq 라이브러리는 random hexamer로 시작해 앞부분 염기 조성이 치우칩니다. 알려진 정상 현상 → 굳이 안 자릅니다.
- Sequence duplication / Overrepresented 높음: 많이 발현된 유전자는 같은 read를 대량 생산합니다. 그건 진짜 생물학적 신호지 오류가 아닙니다(DNA-seq이면 문제, RNA-seq은 기대되는 것).
→ 신호등 색만 보지 말고 “RNA-seq라서 그런 것” 인지 구분해야 합니다.
QC 결과 → 다음 행동
섹션 제목: “QC 결과 → 다음 행동”어댑터 함량 높음 → 트리밍에서 어댑터 제거read 끝 품질 낮음 → 끝부분 quality 트리밍전반적으로 좋음 → 가벼운 트리밍 또는 그대로 정렬특정 위치 N/이상 급증 → 장비/레인 문제 의심, 원인 확인실제 리포트 읽기: 항목별 그래프 해석
섹션 제목: “실제 리포트 읽기: 항목별 그래프 해석”아래는 공개 human RNA-seq 샘플(224만 read pair, 100 bp, 트리밍 전)에 FastQC를 돌려 나온 실제 그래프입니다. 각 항목을 어떻게 읽는지 그림과 함께 봅니다.
Per base sequence quality 🟢
섹션 제목: “Per base sequence quality 🟢”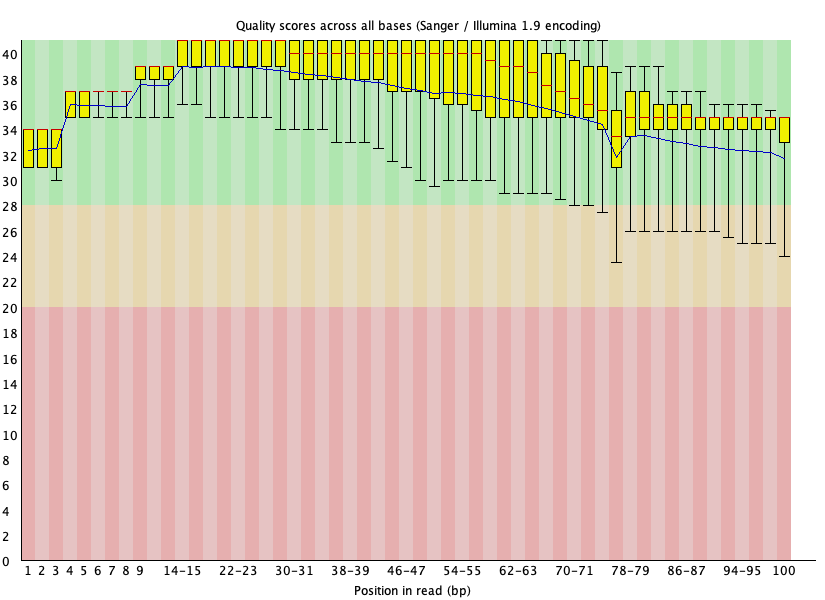
- 읽는 법: x=위치, y=품질. 각 위치의 상자(사분위)와 파란 중앙값 선이 초록(Q28+) 에 있으면 좋음. 상자가 노랑·빨강으로 내려가면 그 위치를 트리밍.
- 이 데이터: 뒤로 갈수록 살짝 낮아지지만 중앙값이 끝(위치 100)까지 Q32대로 초록 유지. → 품질 트리밍 거의 불필요.
Per sequence quality scores 🟢
섹션 제목: “Per sequence quality scores 🟢”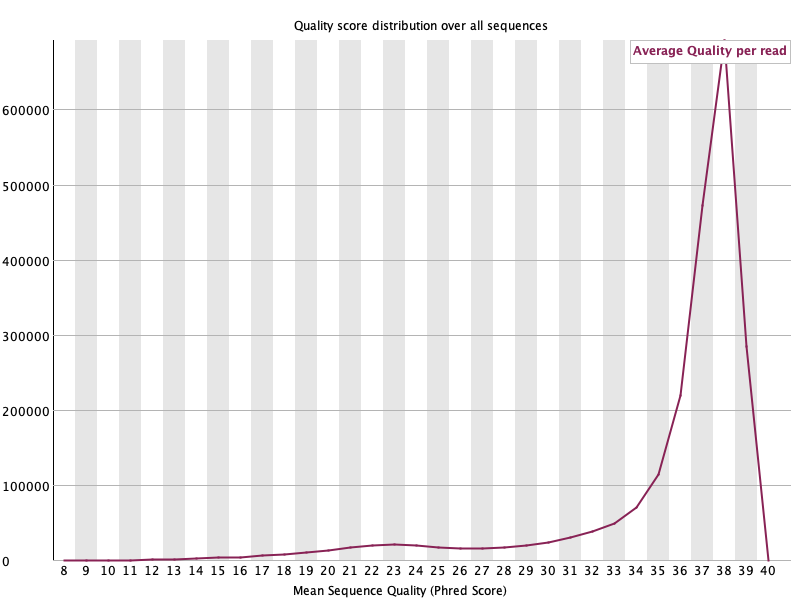
- 읽는 법: read 하나하나의 평균 품질 분포. 높은 Q에 봉우리가 몰릴수록 좋음. 낮은 Q에 별도 봉우리가 있으면 부실한 read 집단이 있다는 뜻.
- 이 데이터: Q38에 날카로운 단일 봉우리: 대부분 read가 고품질. 낮은 쪽 꼬리 미미. 🟢
Per tile sequence quality 🟢
섹션 제목: “Per tile sequence quality 🟢”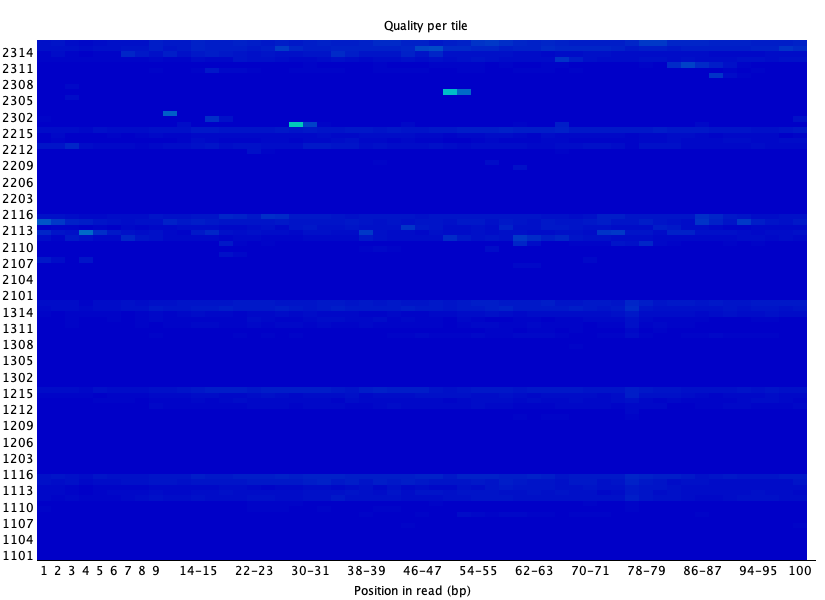
- 읽는 법: flow cell tile(행)×위치(열) 히트맵. 파랑=그 자리 평균 이상, 따뜻한 색=평균 이하. 특정 tile 줄이 빨개지면 그 구획에 기포·먼지 등 물리적 문제. (tile 개념: Flow cell · Lane)
- 이 데이터: 거의 전체가 균일한 파랑, 희미한 점 몇 개뿐 → 물리적 이상 없음. 🟢
Per base sequence content 🔴 (정상)
섹션 제목: “Per base sequence content 🔴 (정상)”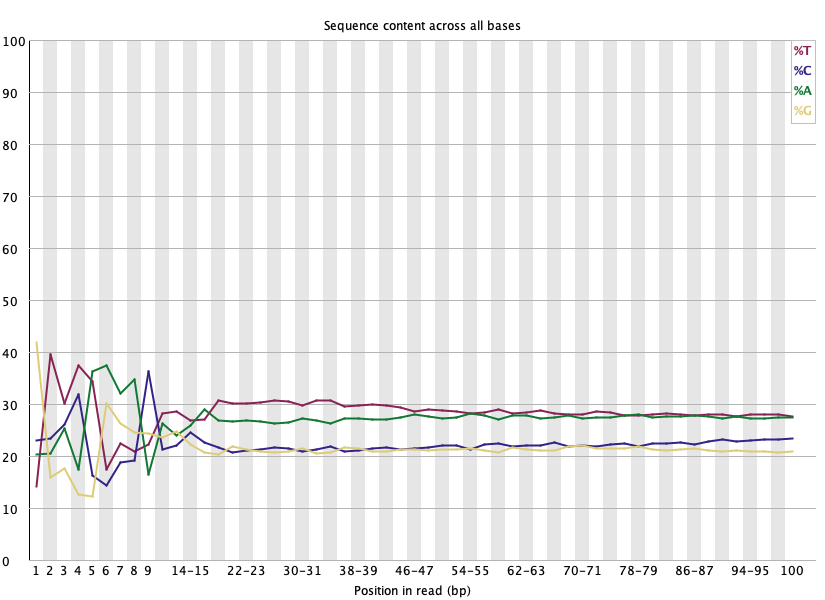
- 읽는 법: 위치별 4염기 비율. 이론상 평평해야 하고, 위치별로 크게 갈리면 FAIL.
- 이 데이터: 앞 ~12 bp가 요동치다가 이후 평평. FastQC는 이걸 FAIL로 표시하지만: RNA-seq의 random hexamer 프라이밍 편향으로 정상입니다. 손대지 않음.
Per sequence GC content 🟢
섹션 제목: “Per sequence GC content 🟢”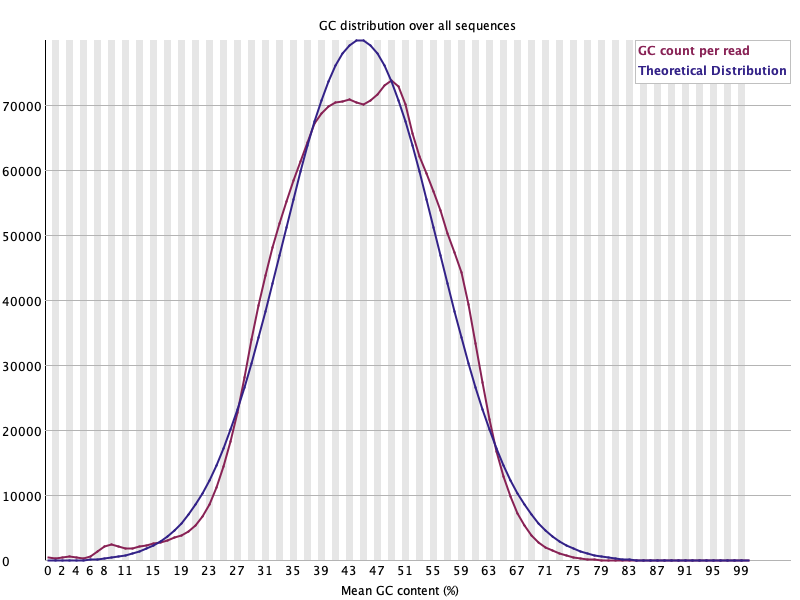
- 읽는 법: read별 GC% 분포(빨강)가 이론 정규분포(파랑)를 따르는지. 예상 밖 봉우리는 오염(다른 생물·rRNA·어댑터) 신호.
- 이 데이터: 관측이 이론 곡선을 잘 따라가고 피크 ~45%. 살짝 어깨가 있지만 큰 이중 봉우리 없음 → 오염 징후 없음. 🟢
Per base N content 🟢
섹션 제목: “Per base N content 🟢”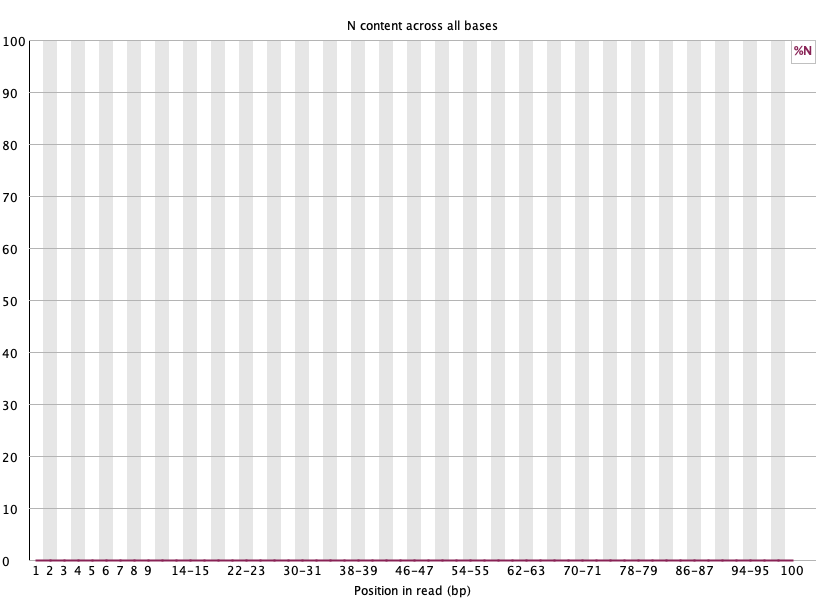
- 읽는 법: 위치별 N(판독 실패) 비율. 0에 붙어 있어야 정상. 특정 위치서 치솟으면 장비/화학 문제.
- 이 데이터: 전 구간 0 → 문제 없음. 🟢
Sequence Length Distribution 🟢
섹션 제목: “Sequence Length Distribution 🟢”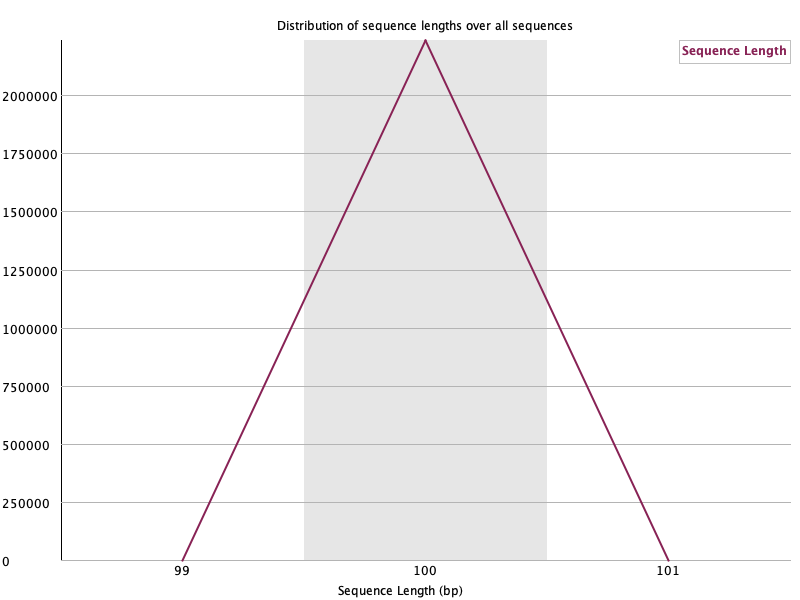
- 읽는 법: read 길이 분포. 트리밍 전이면 대개 한 값에 몰림.
- 이 데이터: 100 bp에 단일 스파이크(모든 read가 100 bp). 🟢 트리밍 후엔 길이가 퍼져 WARN이 뜨는데, 그건 정상입니다.
Sequence Duplication Levels 🟢
섹션 제목: “Sequence Duplication Levels 🟢”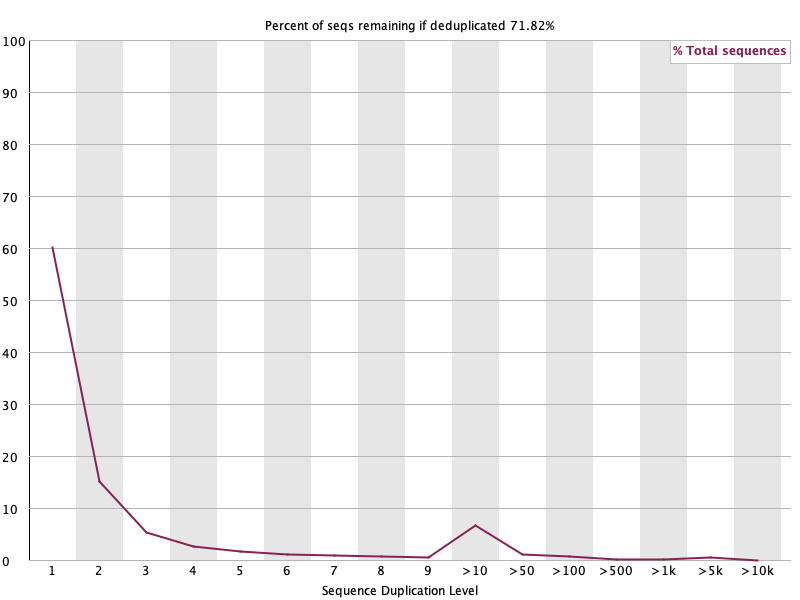
- 읽는 법: 같은 서열이 몇 번 중복되는지. 제목의 ”% remaining if deduplicated” 가 높을수록 중복이 적음.
- 이 데이터: 제거 시 71.82% 잔존(≈28% 중복), 대부분 고유(level 1).
>10의 작은 봉우리는 고발현 유전자·polyA 꼬리가 만든 것: RNA-seq에선 정상. 🟢
Adapter Content 🟡
섹션 제목: “Adapter Content 🟡”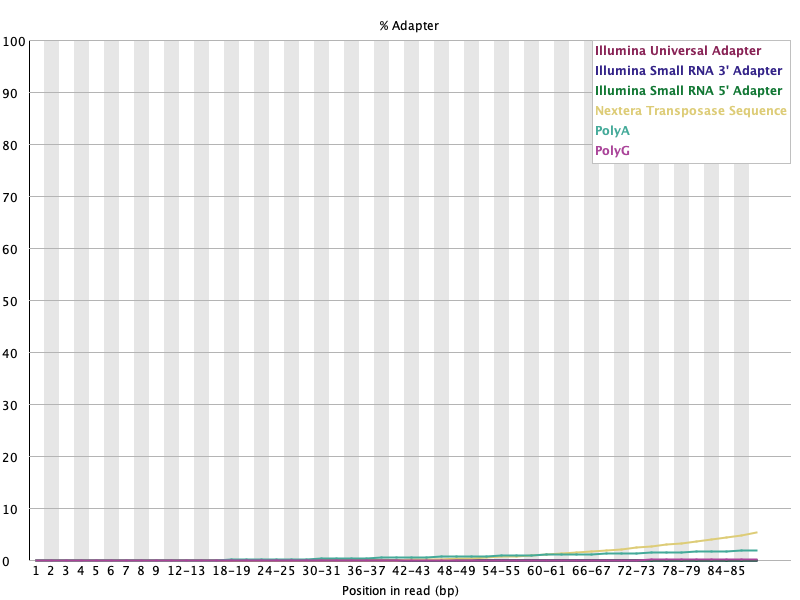
- 읽는 법: 위치별 어댑터 서열 누적 함량. 끝으로 갈수록 오르면 read-through된 어댑터가 있다는 뜻.
- 이 데이터: 대부분 0 근처, read 끝(~88 bp)에서 Nextera(노란선)가 ~5%로 상승 → WARN. 어댑터 트리밍의 직접 근거. 🟡
종합 판단
섹션 제목: “종합 판단”- 품질은 훌륭(Per base/ per sequence quality 모두 🟢) → quality 트리밍은 최소.
- FAIL·WARN은 Per base content(RNA-seq 정상)와 Adapter content(끝 어댑터 소량)뿐.
- → 어댑터·polyA 정리 위주로 가볍게 트리밍하면 충분하다는 결론.
트리밍 후 다시: before/after 검증
섹션 제목: “트리밍 후 다시: before/after 검증”FastQC는 한 번만 돌리는 게 아닙니다. 표준 흐름은 QC → 트리밍 → QC 다시입니다.
FASTQ ──FastQC(before)──▶ 문제 진단 ──트리밍(fastp)───▶ 정리된 FASTQ ──FastQC(after)───▶ 정말 나아졌나 확인 ──▶ 정렬로트리밍을 한 뒤 다시 FastQC를 돌리는 이유:
- 어댑터가 실제로 제거됐나 확인 (WARN이 사라져야 함)
- 품질이 유지/개선됐나 확인 (나빠지면 안 됨)
- 과하게 자르지 않았나 확인 (너무 짧아진 read·이상 신호)
실제 before/after: 신호등 변화
섹션 제목: “실제 before/after: 신호등 변화”같은 샘플을 트리밍 전/후로 FastQC 비교:
| 항목 | before | after | 해석 |
|---|---|---|---|
| Adapter Content | 🟡 WARN | 🟢 PASS | 어댑터 제거됨: 트리밍의 목적 달성 ✅ |
| Sequence Length Distribution | 🟢 PASS | 🟡 WARN | 길이가 다양해짐: 트리밍하면 당연, 정상 |
| Per base sequence content | 🔴 FAIL | 🔴 FAIL | random hexamer(생물학적) → 트리밍으로 안 변함 |
| Per base/sequence quality | 🟢 | 🟢 | 품질 유지 |
→ 요점: 좋아져야 할 것(어댑터)은 좋아지고, 나빠지면 안 되는 것(품질)은 그대로. 새로 뜬 Length WARN은 “트리밍했다”는 흔적일 뿐입니다.
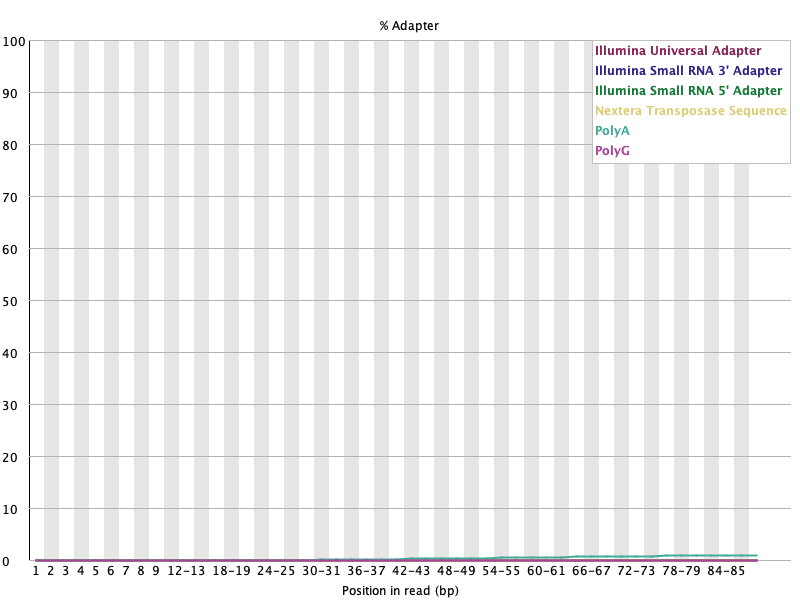
어댑터: WARN이 사라졌다
섹션 제목: “어댑터: WARN이 사라졌다”Before (끝에서 Nextera가 ~5%로 상승):
After (거의 0으로 평평):
길이 분포: WARN이 새로 떴지만 정상
섹션 제목: “길이 분포: WARN이 새로 떴지만 정상”Before (모두 100 bp, 단일 스파이크) → After (짧아진 read가 생겨 분포가 퍼짐):
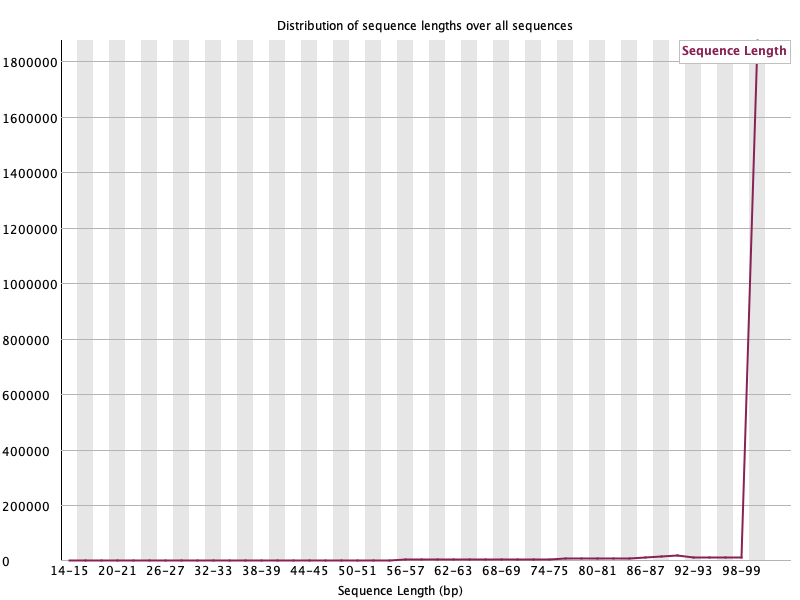
이 WARN은 문제 신호가 아니라 트리밍이 작동했다는 증거입니다. FastQC의 신호등은 맥락으로 읽어야 한다는 또 하나의 예시입니다.
여러 샘플이면: MultiQC
섹션 제목: “여러 샘플이면: MultiQC”샘플이 많아지면 리포트가 흩어집니다. MultiQC가 여러 FastQC 결과(그리고 정렬·정량 로그까지)를 한 페이지로 합쳐 줍니다.
대안·보완 도구
섹션 제목: “대안·보완 도구”FastQC는 2010년 이후 사실상 표준이지만, 그동안 더 빠르거나 특정 상황에 맞는 도구들이 나왔습니다. 역할별로 정리합니다.
드롭인 대체: 더 빠르고 결과는 (거의) 동일
섹션 제목: “드롭인 대체: 더 빠르고 결과는 (거의) 동일”| 도구 | 특징 |
|---|---|
| Falco | FastQC를 C++로 재구현한 드롭인 교체. 평균 ~3배 빠르고 메모리도 적게 쓰며, 출력·모듈이 거의 동일. FastQC(Java)가 느리거나 대용량일 때 1순위 대안. |
| Sequali | 최신 도구(C+Python). 빠르고, 과다서열 검사가 read 끝까지 확인, 기대오류 기반 Phred 평균, paired-end insert size 지원. 단축+장기리드 모두 처리. |
QC + 트리밍 통합: 처리하면서 QC까지
섹션 제목: “QC + 트리밍 통합: 처리하면서 QC까지”| 도구 | 특징 |
|---|---|
| fastp | 트리밍 도구지만 QC 요약(HTML)도 함께 냄. 한 번에 진단+정리. 과다서열 추정 방식이 FastQC와 다름(주기적 카운트 vs 앞 10만 read 표본). |
| Trim Galore | cutadapt + FastQC를 감싼 래퍼. |
| HTQC / AfterQC | QC+트리밍 결합형(초기 세대). |
취합: 대체가 아니라 상위 도구
섹션 제목: “취합: 대체가 아니라 상위 도구”| 도구 | 특징 |
|---|---|
| MultiQC | FastQC·fastp·정렬·정량 로그를 한 페이지로 합침. 샘플이 많거나 before/after를 비교할 때 사실상 필수. |
장기리드(나노포어·PacBio): FastQC는 단축리드용
섹션 제목: “장기리드(나노포어·PacBio): FastQC는 단축리드용”| 도구 | 특징 |
|---|---|
| NanoPlot | FASTQ/summary에서 길이·품질 분포 플롯. |
| pycoQC | 나노포어 sequencing_summary.txt 기반 인터랙티브 QC. |
| ToulligQC · LongReadSum | 나노포어 QC·요약. |
이들은 서열 조성·중복·어댑터 오염 진단은 약해서, FastQC류와 성격이 다릅니다.
오염 스크리닝: FastQC가 약한 부분
섹션 제목: “오염 스크리닝: FastQC가 약한 부분”| 도구 | 특징 |
|---|---|
| FastQ Screen | 여러 게놈에 대조해 어느 생물에서 온 read인지(오염·혼입) 확인. |
| Kraken2 | k-mer 기반 분류로 시료 순도 점검. |
어떻게 고르나
섹션 제목: “어떻게 고르나”- 그냥 더 빠른 FastQC가 필요하면 → Falco(드롭인).
- 트리밍이랑 같이 할 거면 → fastp(우리 파이프라인에서 쓰는 것).
- 여러 샘플·리포트를 모아 보려면 → MultiQC.
- 나노포어 장기리드면 → NanoPlot/pycoQC.
- 오염이 의심되면 → FastQ Screen / Kraken2.
- 단, FastQC가 워낙 널리 쓰여 “기준점”으로는 계속 유효합니다.
참고 문헌: Falco (Bioinformatics, 2021) · Sequali (Bioinformatics Advances, 2025)
한 줄 요약: FastQC = read 품질을 진단하는 첫 도구. 데이터를 바꾸지 않고 리포트만 만들며, 그 결과로 트리밍 방법을 정합니다. RNA-seq에선 정상인데 FAIL 뜨는 항목(random hexamer, 고발현 중복)을 구분해 읽는 게 핵심입니다.